

GHB analogs confer neuroprotection through specific interaction with the CaMKIIα hub domain
- PMID: 34330837
- PMCID: PMC8346900
- DOI: 10.1073/pnas.2108079118
GHB analogs confer neuroprotection through specific interaction with the CaMKIIα hub domain
Abstract
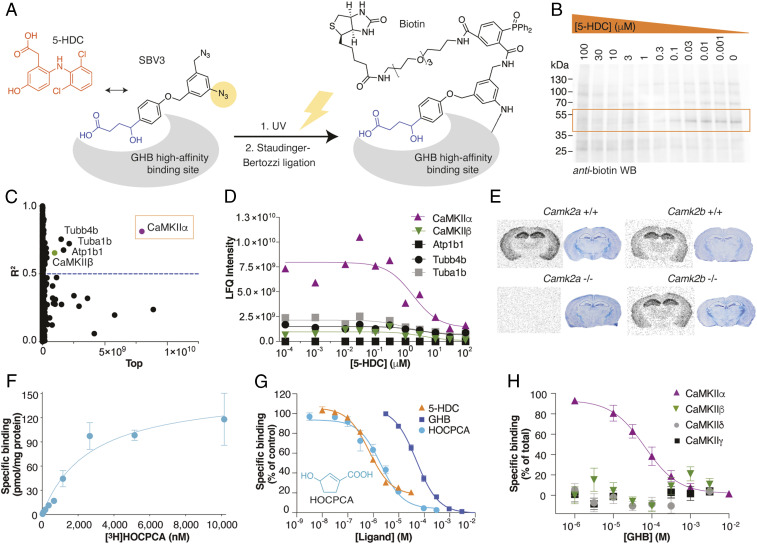
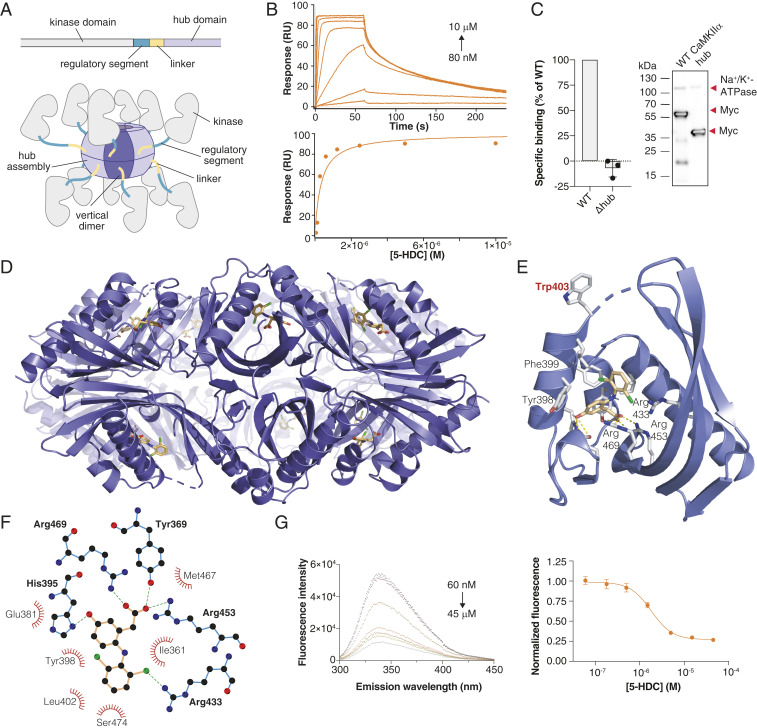
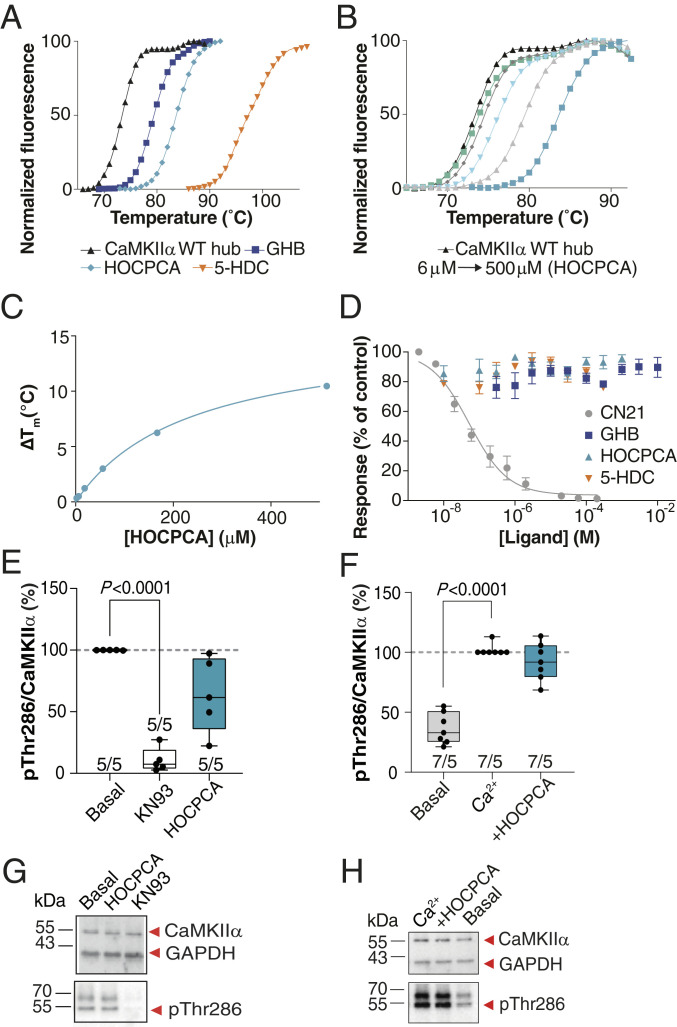
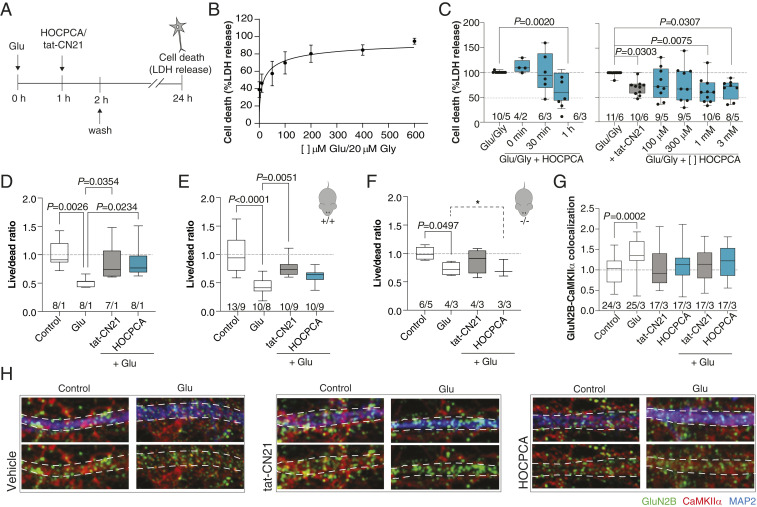
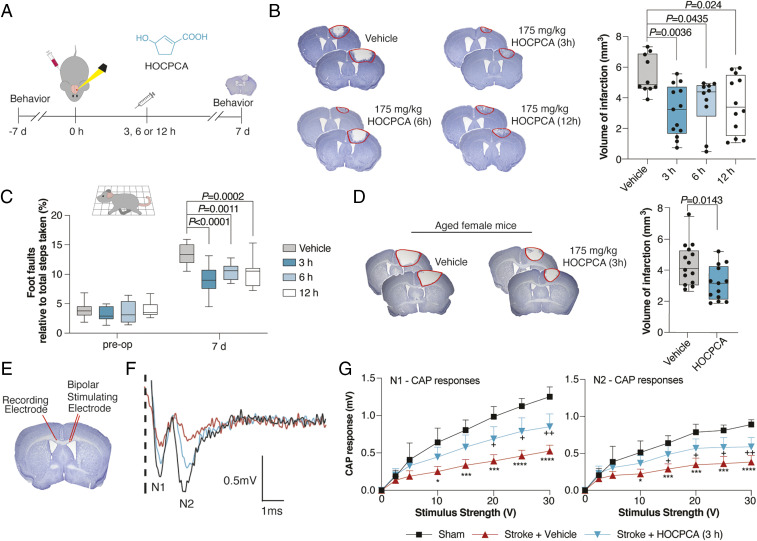
Ca2+/calmodulin-dependent protein kinase II alpha subunit (CaMKIIα) is a key neuronal signaling protein and an emerging drug target. The central hub domain regulates the activity of CaMKIIα by organizing the holoenzyme complex into functional oligomers, yet pharmacological modulation of the hub domain has never been demonstrated. Here, using a combination of photoaffinity labeling and chemical proteomics, we show that compounds related to the natural substance γ-hydroxybutyrate (GHB) bind selectively to CaMKIIα. By means of a 2.2-Å x-ray crystal structure of ligand-bound CaMKIIα hub, we reveal the molecular details of the binding site deep within the hub. Furthermore, we show that binding of GHB and related analogs to this site promotes concentration-dependent increases in hub thermal stability believed to alter holoenzyme functionality. Selectively under states of pathological CaMKIIα activation, hub ligands provide a significant and sustained neuroprotection, which is both time and dose dependent. This is demonstrated in neurons exposed to excitotoxicity and in a mouse model of cerebral ischemia with the selective GHB analog, HOCPCA (3-hydroxycyclopent-1-enecarboxylic acid). Together, our results indicate a hitherto unknown mechanism for neuroprotection by a highly specific and unforeseen interaction between the CaMKIIα hub domain and small molecule brain-penetrant GHB analogs. This establishes GHB analogs as powerful tools for investigating CaMKII neuropharmacology in general and as potential therapeutic compounds for cerebral ischemia in particular.
Keywords: HOCPCA; excitotoxicity; photoaffinity labeling; photothrombotic stroke; x-ray crystallography.
Copyright © 2021 the Author(s). Published by PNAS.
Conflict of interest statement
Competing interest statement: The University of Copenhagen and Otago Innovation Ltd. have licensed the patent rights for GHB derivatives and their uses (WO/2019/149329) to Ceremedy Ltd., of which B.F., B.R.K., and P.W. are cofounders.
Figures
References
-
- De Koninck P., Schulman H., Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 279, 227–230 (1998). - PubMed
-
- Rosenberg O. S., et al. ., Oligomerization states of the association domain and the holoenyzme of Ca2+/CaM kinase II. FEBS J. 273, 682–694 (2006). - PubMed
-
- Hoelz A., Nairn A. C., Kuriyan J., Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol. Cell 11, 1241–1251 (2003). - PubMed
-
- Rosenberg O. S., Deindl S., Sung R. J., Nairn A. C., Kuriyan J., Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell 123, 849–860 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
