

Re-analysis of whole-exome sequencing data reveals a novel splicing variant in the SLC2A1 in a patient with GLUT1 Deficiency Syndrome 1 accompanied by hemangioma: a case report
- PMID: 34332575
- PMCID: PMC8325841
- DOI: 10.1186/s12920-021-01045-3
Re-analysis of whole-exome sequencing data reveals a novel splicing variant in the SLC2A1 in a patient with GLUT1 Deficiency Syndrome 1 accompanied by hemangioma: a case report
Abstract
Background: GLUT1 Deficiency Syndrome 1 (GLUT1DS1) is a neurological disorder caused by either heterozygous or homozygous mutations in the Solute Carrier Family 2, Member 1 (SLC2A1) gene. SLC2A1 encodes Glucose transporter type 1 (GLUT1) protein, which is the primary glucose transporter at the blood-brain barrier. A ketogenic diet (KD) provides an alternative fuel for brain metabolism to treat impaired glucose transport. By reanalyzing exome data, we identified a de novo heterozygous SLC2A1 variant in a girl with epilepsy. After reversed phenotyping with neurometabolic tests, she was diagnosed with GLUT1DS1 and started on a KD. The patient's symptoms responded to the diet. Here, we report a patient with GLUT1DS1 with a novel SLC2A1 mutation. She also has a hemangioma which has not been reported in association with this syndrome before.
Case presentation: A 5-year 8-month girl with global developmental delay, spasticity, intellectual disability, dysarthric speech, abnormal eye movements, and hemangioma. The electroencephalography (EEG) result revealed that she had epilepsy. Magnetic resonance imaging (MRI) showed that non-specific white matter abnormalities. Whole Exome Sequencing (WES) was previously performed, but the case remained unsolved. The re-analysis of WES data revealed a heterozygous splicing variant in the SLC2A1 gene. Segregation analysis with parental DNA samples indicated that the variant occurred de novo. Lumbar puncture (LP) confirmed the diagnosis, and the patient started on a KD. Her seizures responded to the KD. She has been seizure-free since shortly after the initiation of the diet. She also had decreased involuntary movements, her speech became more understandable, and her vocabulary increased after the diet.
Conclusions: We identified a novel de novo variant in the SLC2A1 gene in a patient who previously had a negative WES result. The patient has been diagnosed with GLUT1DS1. The syndrome is a treatable condition, but the differential diagnosis is not an easy process due to showing a wide range of phenotypic spectrum and the overlapping symptoms with other neurological diseases. The diagnosis necessitates a genomic testing approach. Our findings also highlight the importance of re-analysis to undiagnosed cases after initial WES to reveal disease-causing variants.
Keywords: GLUT1 Deficiency Syndrome 1; Hemangioma; Ketogenic diet; SLC2A1; Whole exome sequencing.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
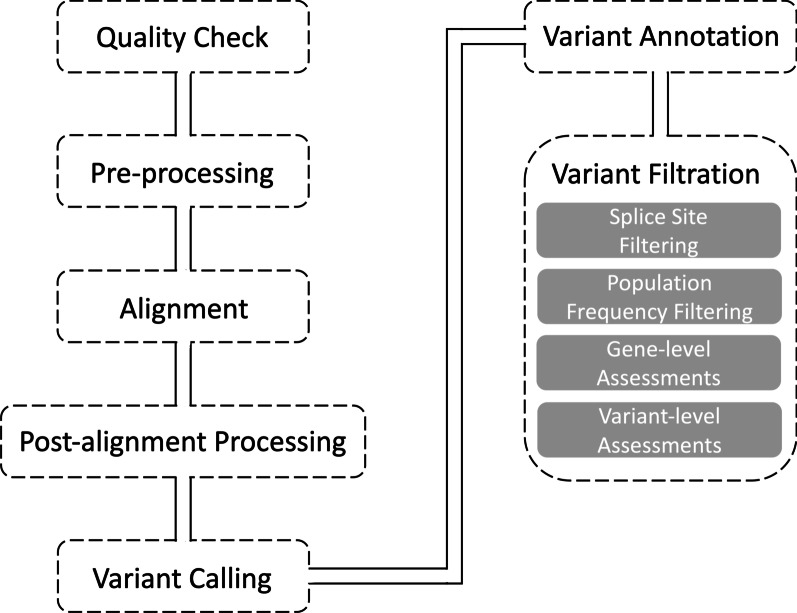

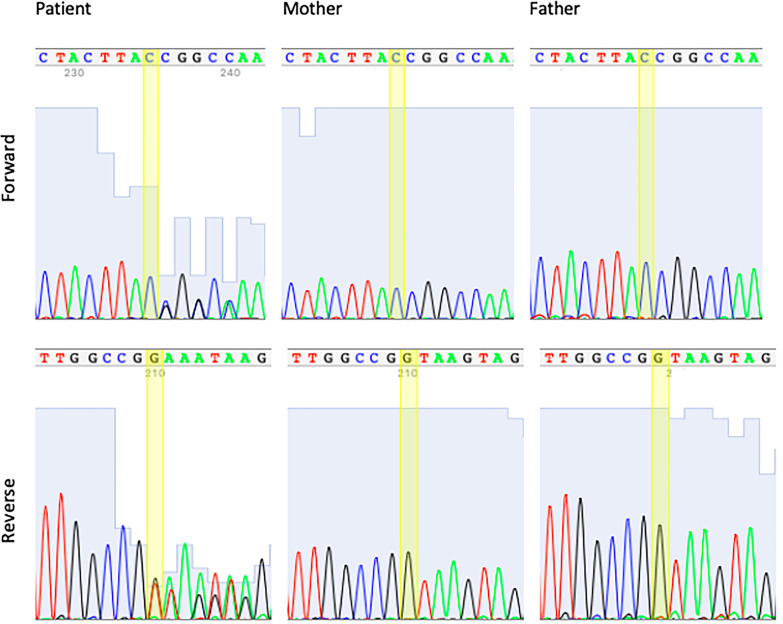
Similar articles
-
[Clinical and genetic characteristics of glucose transporter type 1 deficiency syndrome].Zhonghua Er Ke Za Zhi. 2013 Jun;51(6):443-7. Zhonghua Er Ke Za Zhi. 2013. PMID: 24120063 Chinese.
-
Glucose transporter-1 deficiency syndrome with extreme phenotypic variability in a five-generation family carrying a novel SLC2A1 variant.Eur J Neurol. 2024 Aug;31(8):e16325. doi: 10.1111/ene.16325. Epub 2024 May 27. Eur J Neurol. 2024. PMID: 38803061 Free PMC article.
-
Occurrence of GLUT1 deficiency syndrome in patients treated with ketogenic diet.Epilepsy Behav. 2014 Mar;32:76-8. doi: 10.1016/j.yebeh.2014.01.003. Epub 2014 Feb 6. Epilepsy Behav. 2014. PMID: 24508593
-
GLUT1 Deficiency Syndrome-Early Treatment Maintains Cognitive Development? (Literature Review and Case Report).Genes (Basel). 2021 Aug 31;12(9):1379. doi: 10.3390/genes12091379. Genes (Basel). 2021. PMID: 34573360 Free PMC article. Review.
-
GLUT1 deficiency syndrome 2013: current state of the art.Seizure. 2013 Dec;22(10):803-11. doi: 10.1016/j.seizure.2013.07.003. Epub 2013 Jul 26. Seizure. 2013. PMID: 23890838 Review.
Cited by
-
Evaluation of the etiology of epilepsy and/or developmental delay in children via next-generation sequencing: a single-center experience.Front Pediatr. 2025 Feb 27;13:1471965. doi: 10.3389/fped.2025.1471965. eCollection 2025. Front Pediatr. 2025. PMID: 40083435 Free PMC article.
-
Dietary management and access to treatment for patients with glucose deficiency syndrome type 1: an overview review with focus on the European regulatory framework.Eur J Clin Nutr. 2024 Dec;78(12):1058-1063. doi: 10.1038/s41430-024-01490-0. Epub 2024 Aug 10. Eur J Clin Nutr. 2024. PMID: 39127841 Review.
-
One Molecule for Mental Nourishment and More: Glucose Transporter Type 1-Biology and Deficiency Syndrome.Biomedicines. 2022 May 26;10(6):1249. doi: 10.3390/biomedicines10061249. Biomedicines. 2022. PMID: 35740271 Free PMC article. Review.
References
-
- Baldwin SA. Mammalian passive glucose transporters: members of an ubiquitous family of active and passive transport proteins. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes. 1993;1154:17–49. - PubMed
-
- Pardridges M, Boado J, Farrell R. Brain-type glucose transporter (GLUT-1) is selectively localized to the blood-brain barrier. J Biol. 1990;265:18035–18040. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
