

Membrane-Facilitated Receptor Access and Binding Mechanisms of Long-Acting β 2-Adrenergic Receptor Agonists
- PMID: 34334369
- PMCID: PMC8626642
- DOI: 10.1124/molpharm.121.000285
Membrane-Facilitated Receptor Access and Binding Mechanisms of Long-Acting β 2-Adrenergic Receptor Agonists
Abstract
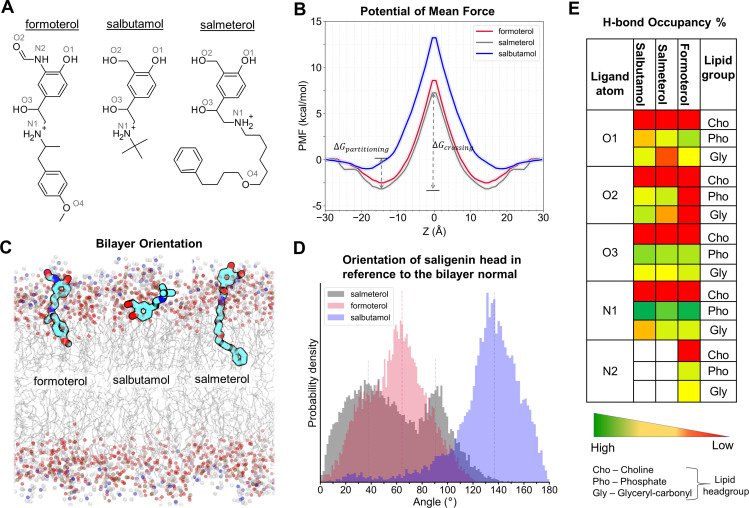
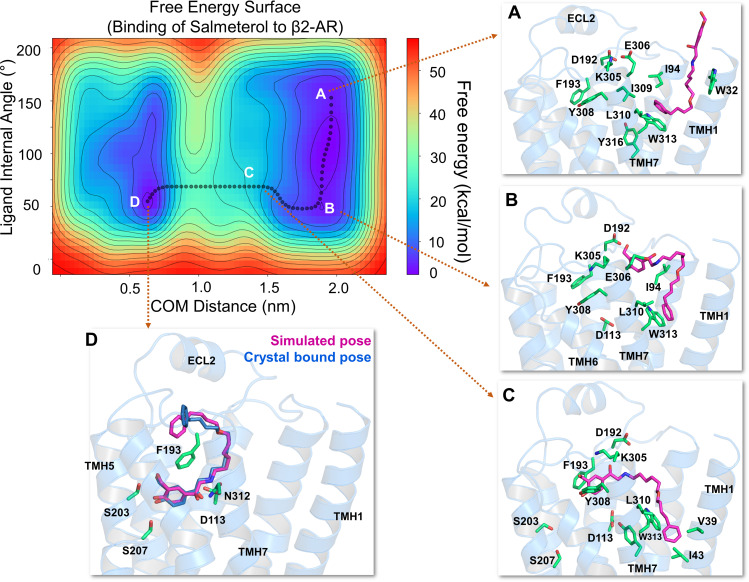
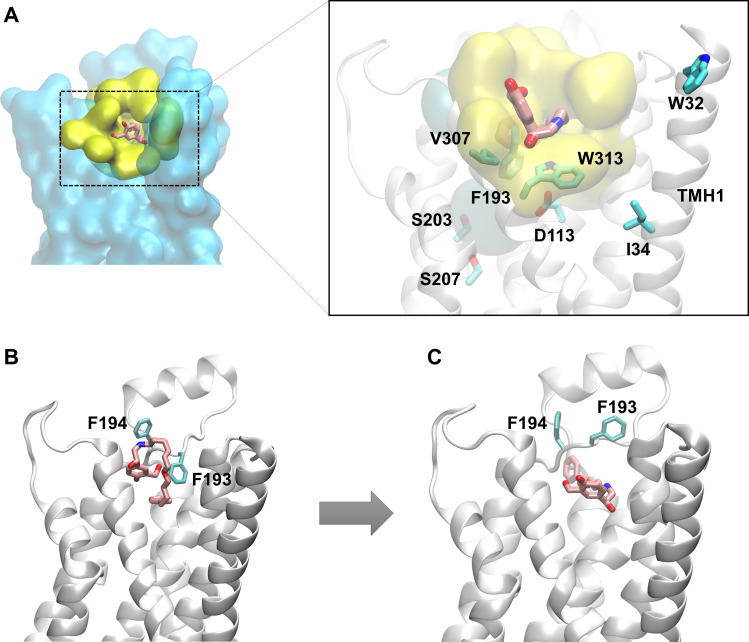
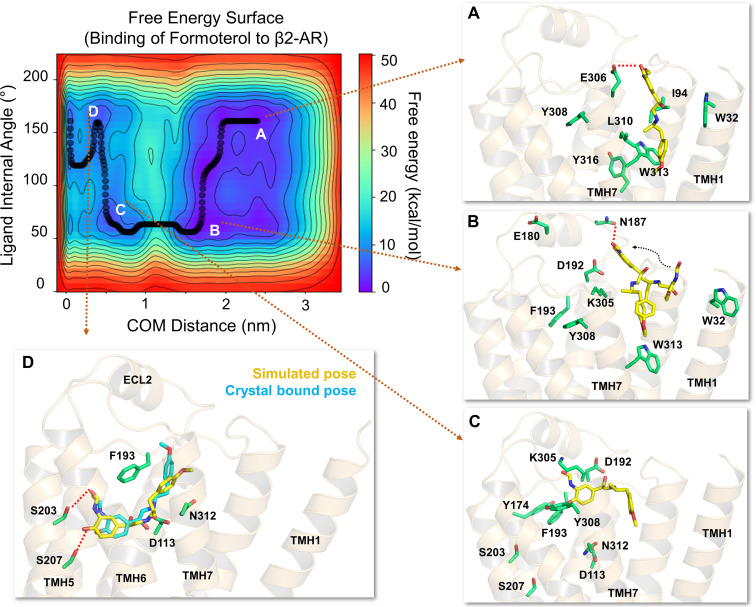
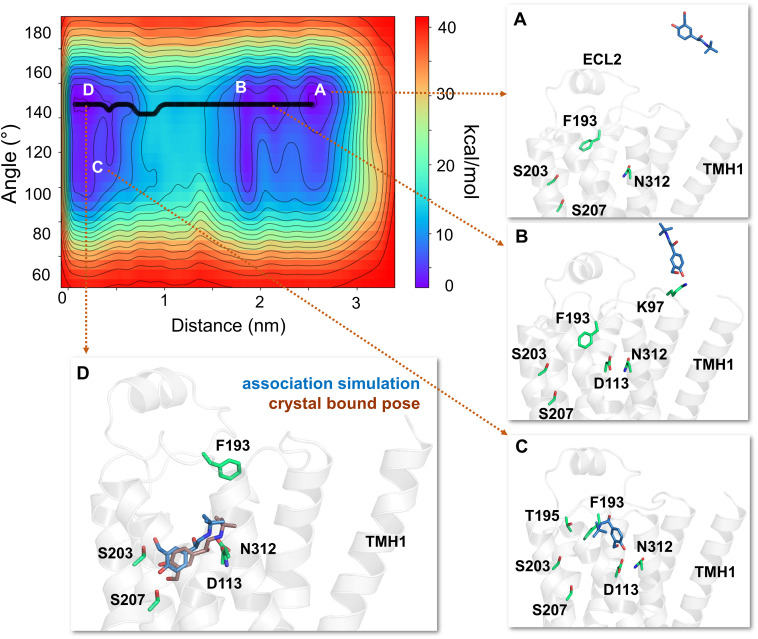
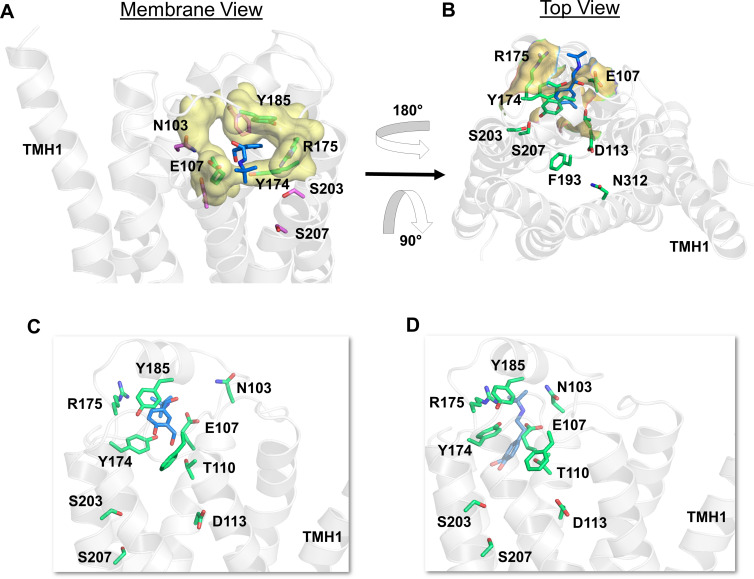
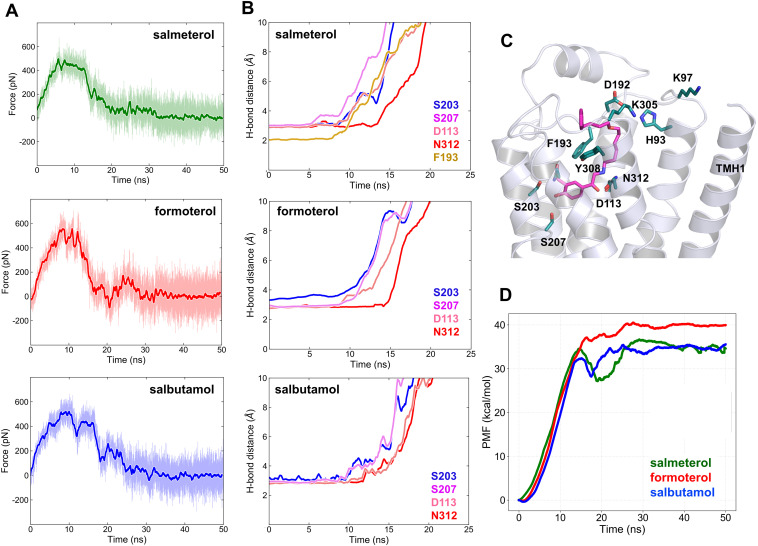
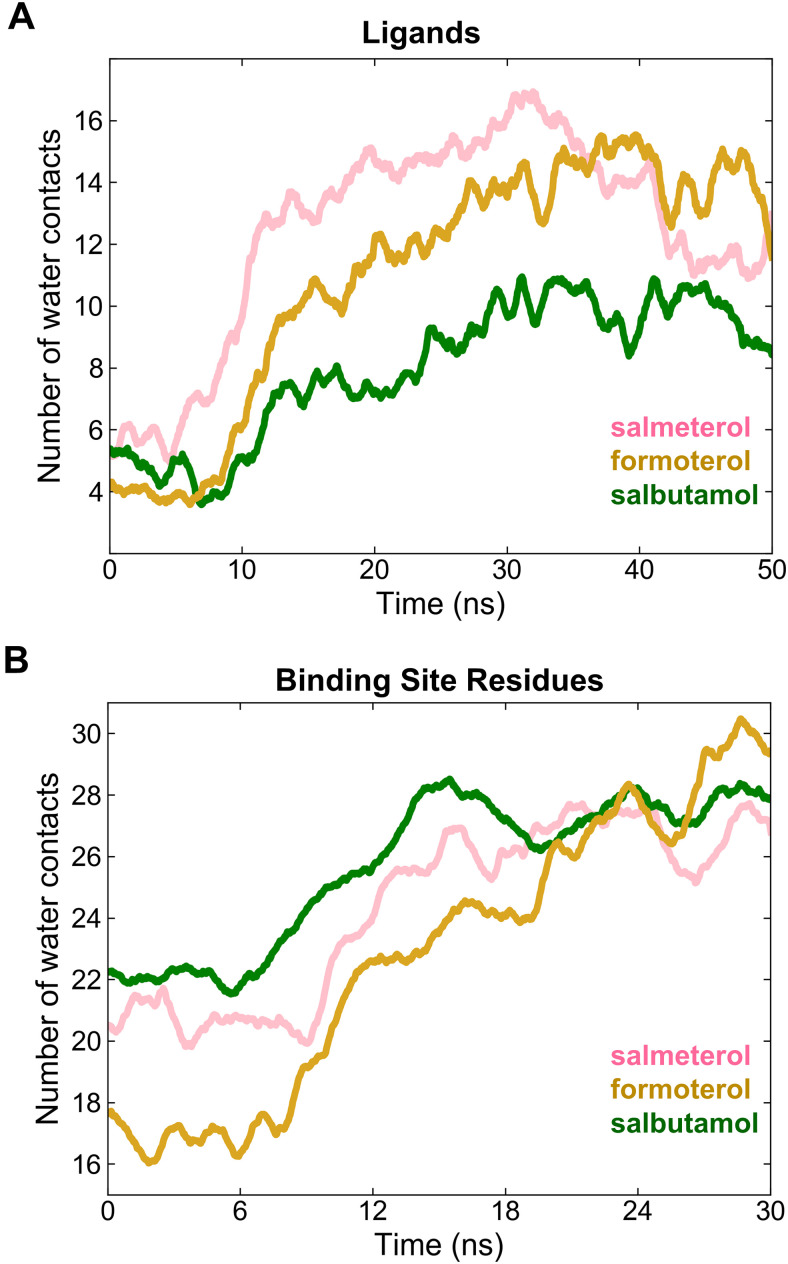
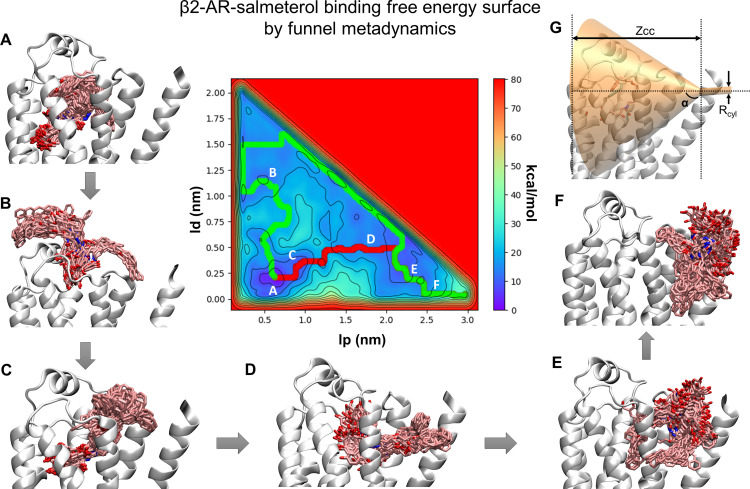
The drugs salmeterol, formoterol, and salbutamol constitute the frontline treatment of asthma and other chronic pulmonary diseases. These drugs activate the β2-adrenergic receptors (β2-AR), a class A G protein-coupled receptor (GPCR), and differ significantly in their clinical onset and duration of actions. According to the microkinetic model, the long duration of action of salmeterol and formoterol compared with salbutamol were attributed, at least in part, to their high lipophilicity and increased local concentrations in the membrane near the receptor. However, the structural and molecular bases of how the lipophilic drugs reach the binding site of the receptor from the surrounding membrane remain unknown. Using a variety of classic and enhanced molecular dynamics simulation techniques, we investigated the membrane partitioning characteristics, binding, and unbinding mechanisms of the ligands. The obtained results offer remarkable insight into the functional role of membrane lipids in the ligand association process. Strikingly, salmeterol entered the binding site from the bilayer through transmembrane helices 1 and 7. The entry was preceded by membrane-facilitated rearrangement and presentation of its phenyl-alkoxy-alkyl tail as a passkey to an access route gated by F193, a residue known to be critical for salmeterol's affinity. Formoterol's access is through the aqueous path shared by other β2-AR agents. We observed a novel secondary path for salbutamol that is distinct from its primary route. Our study offers a mechanistic description for the membrane-facilitated access and binding of ligands to a membrane protein and establishes a groundwork for recognizing membrane lipids as an integral component in the molecular recognition process. SIGNIFICANCE STATEMENT: The cell membrane's functional role behind the duration of action of long-acting β2-adrenergic receptor (β2-AR) agonists such as salmeterol has been a subject of debate for a long time. This study investigated the binding and unbinding mechanisms of the three commonly used β2-AR agonists, salmeterol, formoterol, and salbutamol, using advanced simulation techniques. The obtained results offer unprecedented insights into the active role of membrane lipids in facilitating access and binding of the ligands, affecting the molecular recognition process and thus their pharmacology.
Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics.
Figures

Similar articles
-
Salmeterol's extreme β2 selectivity is due to residues in both extracellular loops and transmembrane domains.Mol Pharmacol. 2015 Jan;87(1):103-20. doi: 10.1124/mol.114.095364. Epub 2014 Oct 16. Mol Pharmacol. 2015. PMID: 25324048
-
Pharmacological similarities and differences between beta2-agonists.Respir Med. 2001 Aug;95 Suppl B:S7-11. doi: 10.1053/rmed.2001.1139. Respir Med. 2001. PMID: 11534896 Review.
-
Beta-2 Adrenergic Agonists Are Substrates and Inhibitors of Human Organic Cation Transporter 1.Mol Pharm. 2015 Aug 3;12(8):2633-41. doi: 10.1021/mp500854e. Epub 2015 Mar 18. Mol Pharm. 2015. PMID: 25751092
-
Decreased bronchodilating effect of salbutamol in relieving methacholine induced moderate to severe bronchoconstriction during high dose treatment with long acting beta2 agonists.Thorax. 2001 Jul;56(7):529-35. doi: 10.1136/thorax.56.7.529. Thorax. 2001. PMID: 11413351 Free PMC article. Clinical Trial.
-
Why are long-acting beta-adrenoceptor agonists long-acting?Eur Respir J. 1994 Mar;7(3):569-78. doi: 10.1183/09031936.94.07030569. Eur Respir J. 1994. PMID: 7912202 Review.
Cited by
-
Molecular basis for the recognition of 24-(S)-hydroxycholesterol by integrin αvβ3.Sci Rep. 2023 Jun 6;13(1):9166. doi: 10.1038/s41598-023-36040-4. Sci Rep. 2023. PMID: 37280310 Free PMC article.
-
Membrane Lipids Are an Integral Part of Transmembrane Allosteric Sites in GPCRs: A Case Study of Cannabinoid CB1 Receptor Bound to a Negative Allosteric Modulator, ORG27569, and Analogs.J Med Chem. 2022 Sep 22;65(18):12240-12255. doi: 10.1021/acs.jmedchem.2c00946. Epub 2022 Sep 6. J Med Chem. 2022. PMID: 36066412 Free PMC article.
-
Characterization of a novel positive allosteric modulator of the α1A-Adrenergic receptor.Curr Res Pharmacol Drug Discov. 2022 Dec 2;4:100142. doi: 10.1016/j.crphar.2022.100142. eCollection 2023. Curr Res Pharmacol Drug Discov. 2022. PMID: 36544813 Free PMC article.
-
Large scale investigation of GPCR molecular dynamics data uncovers allosteric sites and lateral gateways.Nat Commun. 2025 Feb 27;16(1):2020. doi: 10.1038/s41467-025-57034-y. Nat Commun. 2025. PMID: 40016203 Free PMC article.
-
Allosteric modulation of α1β3γ2 GABAA receptors by farnesol through the neurosteroid sites.Biophys J. 2023 Mar 7;122(5):849-867. doi: 10.1016/j.bpj.2023.01.032. Epub 2023 Jan 31. Biophys J. 2023. PMID: 36721367 Free PMC article.
References
-
- (2019)Molecular Operating Environment (MOE), 2013.08, Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada.
-
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25.
-
- Anderson GP, Lindén A, Rabe KF (1994) Why are long-acting beta-adrenoceptor agonists long-acting? Eur Respir J 7:569–578. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
