

S-Nitrosylation of RhoGAP Myosin9A Is Altered in Advanced Diabetic Kidney Disease
- PMID: 34336885
- PMCID: PMC8316719
- DOI: 10.3389/fmed.2021.679518
S-Nitrosylation of RhoGAP Myosin9A Is Altered in Advanced Diabetic Kidney Disease
Abstract
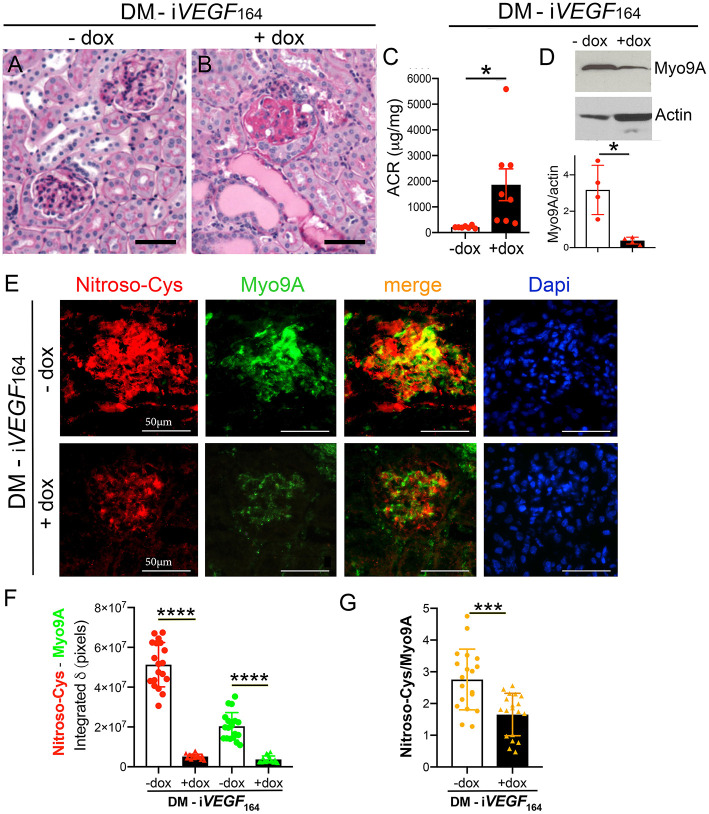
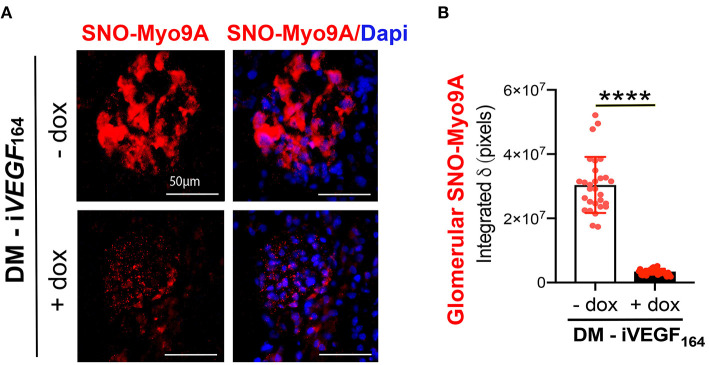
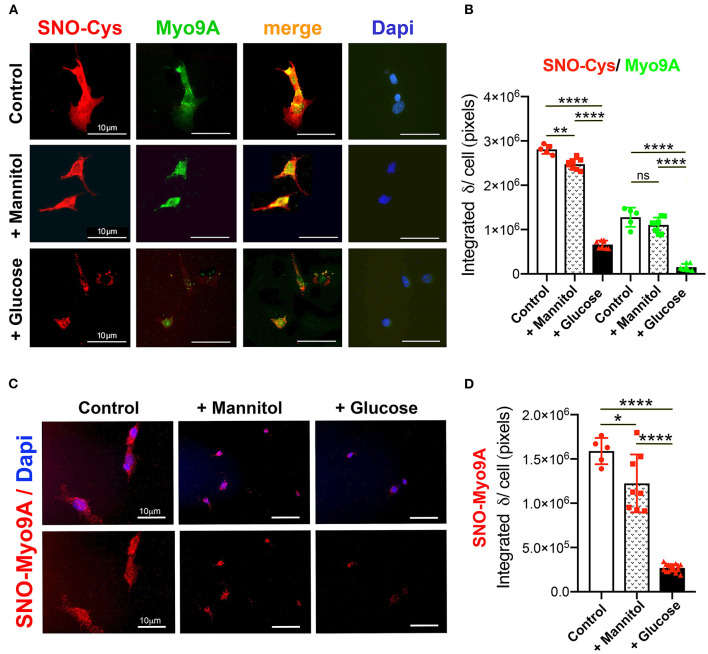
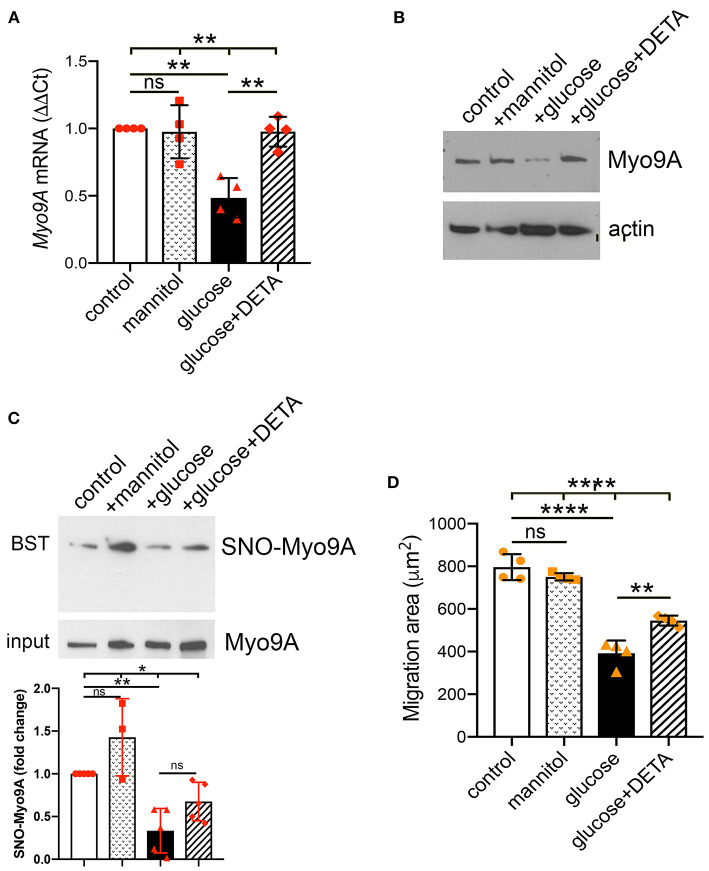
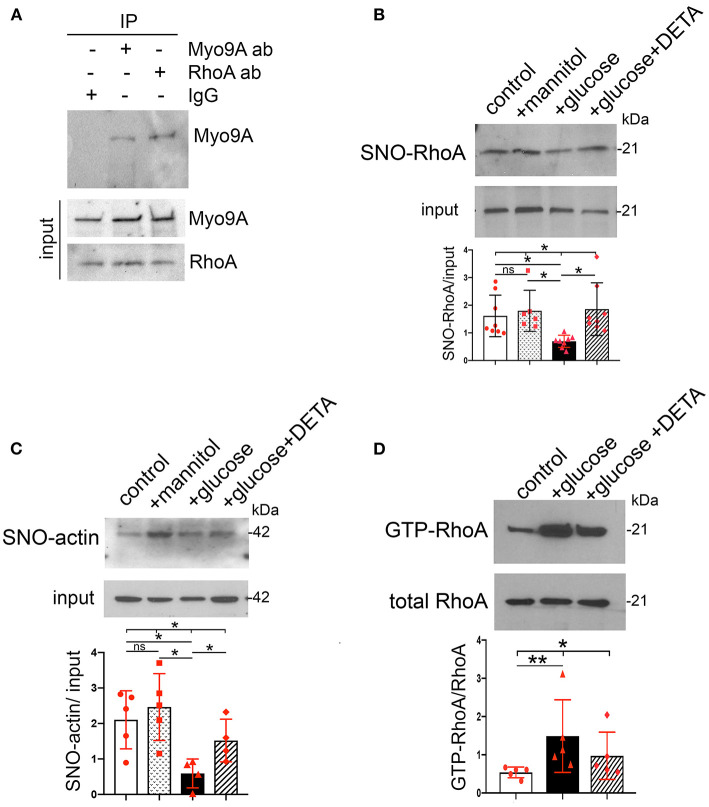
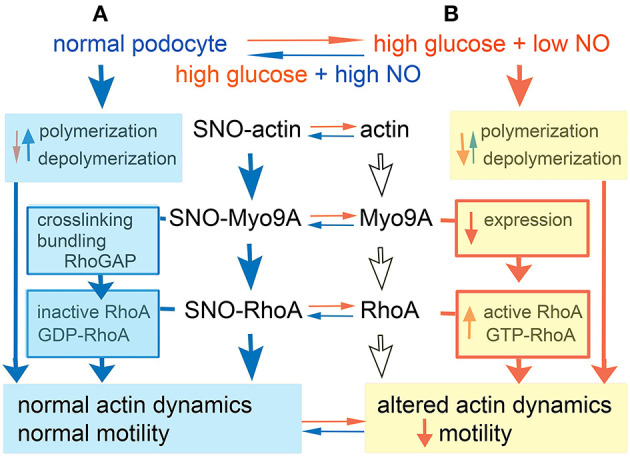
The molecular pathogenesis of diabetic kidney disease progression is complex and remains unresolved. Rho-GAP MYO9A was recently identified as a novel podocyte protein and a candidate gene for monogenic FSGS. Myo9A involvement in diabetic kidney disease has been suggested. Here, we examined the effect of diabetic milieu on Myo9A expression in vivo and in vitro. We determined that Myo9A undergoes S-nitrosylation, a post-translational modification dependent on nitric oxide (NO) availability. Diabetic mice with nodular glomerulosclerosis and severe proteinuria associated with doxycycline-induced, podocyte-specific VEGF 164 gain-of-function showed markedly decreased glomerular Myo9A expression and S-nitrosylation, as compared to uninduced diabetic mice. Immortalized mouse podocytes exposed to high glucose revealed decreased Myo9A expression, assessed by qPCR, immunoblot and immunocytochemistry, and reduced Myo9A S-nitrosylation (SNO-Myo9A), assessed by proximity link assay and biotin switch test, functionally resulting in abnormal podocyte migration. These defects were abrogated by exposure to a NO donor and were not due to hyperosmolarity. Our data demonstrate that high-glucose induced decrease of both Myo9A expression and SNO-Myo9A is regulated by NO availability. We detected S-nitrosylation of Myo9A interacting proteins RhoA and actin, which was also altered by high glucose and NO dependent. RhoA activity inversely related to SNO-RhoA. Collectively, data suggest that dysregulation of SNO-Myo9A, SNO-RhoA and SNO-actin may contribute to the pathogenesis of advanced diabetic kidney disease and may be amenable to therapeutic targeting.
Keywords: MYO9A; RhoA; S-nitrosylation; actin; cell cross-talk; diabetic kidney disease.
Copyright © 2021 Li, Veron and Tufro.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous