

Modeling Sporadic Alzheimer's Disease in Human Brain Organoids under Serum Exposure
- PMID: 34337898
- PMCID: PMC8456220
- DOI: 10.1002/advs.202101462
Modeling Sporadic Alzheimer's Disease in Human Brain Organoids under Serum Exposure
Abstract
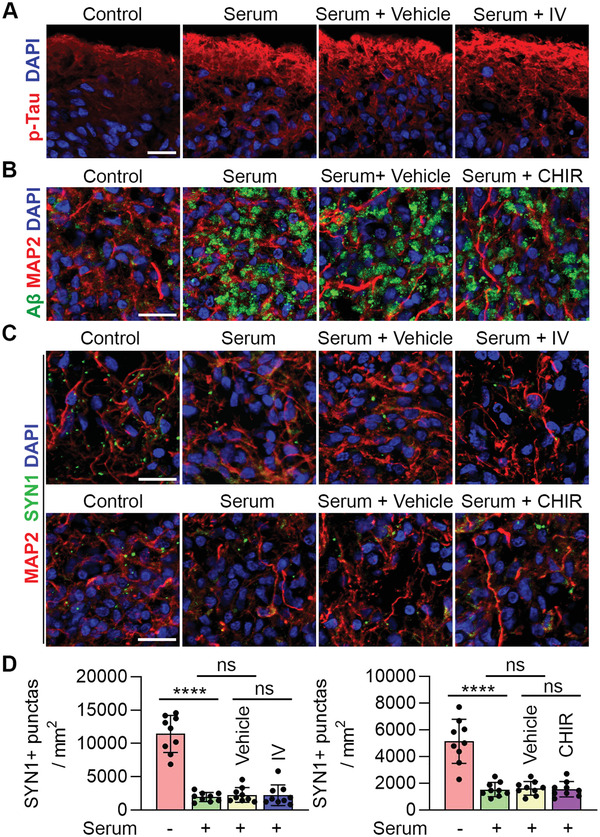
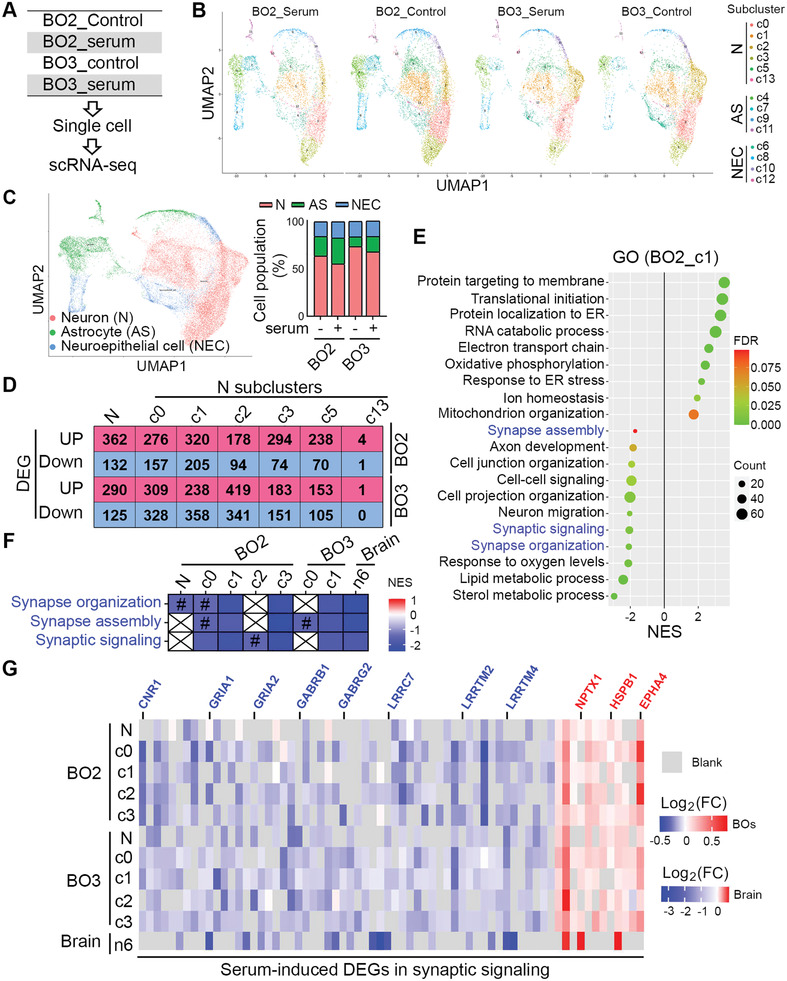
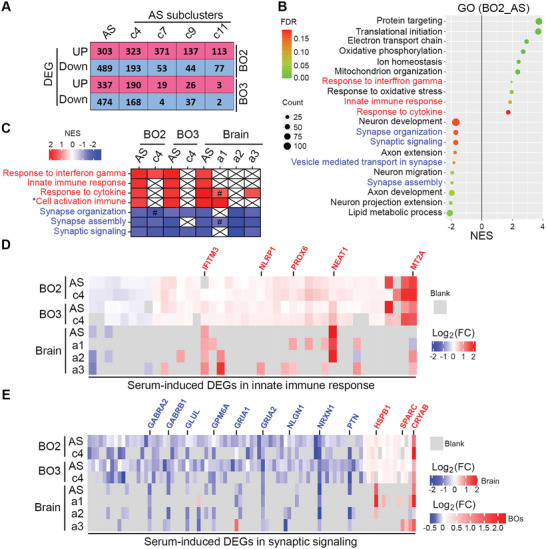
Alzheimer's disease (AD) is a progressive neurodegenerative disease with no cure. Huge efforts have been made to develop anti-AD drugs in the past decades. However, all drug development programs for disease-modifying therapies have failed. Possible reasons for the high failure rate include incomplete understanding of complex pathophysiology of AD, especially sporadic AD (sAD), and species difference between humans and animal models used in preclinical studies. In this study, sAD is modeled using human induced pluripotent stem cell (hiPSC)-derived 3D brain organoids. Because the blood-brain barrier (BBB) leakage is a well-known risk factor for AD, brain organoids are exposed to human serum to mimic the serum exposure consequence of BBB breakdown in AD patient brains. The serum-exposed brain organoids are able to recapitulate AD-like pathologies, including increased amyloid beta (Aβ) aggregates and phosphorylated microtubule-associated tau protein (p-Tau) level, synaptic loss, and impaired neural network. Serum exposure increases Aβ and p-Tau levels through inducing beta-secretase 1 (BACE) and glycogen synthase kinase-3 alpha / beta (GSK3α/β) levels, respectively. In addition, single-cell transcriptomic analysis of brain organoids reveals that serum exposure reduced synaptic function in both neurons and astrocytes and induced immune response in astrocytes. The human brain organoid-based sAD model established in this study can provide a powerful platform for both mechanistic study and therapeutic development in the future.
Keywords: brain organoids; disease modeling; induced pluripotent stem cells; serum exposure; sporadic Alzheimer's disease.
© 2021 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
T.G.B. has contract research with Avid Radiopharmaceuticals. E.M.R. is a scientific advisor to Alkahest, Alzheon, Aural Analytics, Denali, Green Valley, MagQ, Takeda and United Neuroscience. E.M.R. is an advisor to Roche/Roche Diagnostics and Cerveaux (expenses only). E.M.R. is co‐founder and shareholder of AlzPath, a new company which aims to advance the role of blood‐based biomarkers for Alzheimer's disease research, drug development and care. M.B.J. is a co‐inventor of patent WO/2018/160496 related to differentiation of human pluripotent stem cells into microglia. D.M.H. is as an inventor on a patent licensed by Washington University to C2N Diagnostics on the therapeutic use of anti‐tau antibodies. D.M.H. co‐founded and is on the scientific advisory board of C2N Diagnostics. C2N Diagnostics has licensed certain anti‐tau antibodies to AbbVie for therapeutic development. D.M.H. is on the scientific advisory board of Denali and consults for Genentech, Merck, and Cajal Neuroscience.
Figures
References
-
- Gaugler J., James B., Johnson T., Marin A., Weuve J., Assoc A. s., Alzheimers Dementia 2019, 15, 321.
-
- a) Liu C. C., Liu C. C., Kanekiyo T., Xu H., Bu G., Nat. Rev. Neurol. 2013, 9, 106; - PMC - PubMed
- b) Campion D., Dumanchin C., Hannequin D., Dubois B., Belliard S., Puel M., Thomas‐Anterion C., Michon A., Martin C., Charbonnier F., Raux G., Camuzat A., Penet C., Mesnage V., Martinez M., Clerget‐Darpoux F., Brice A., Frebourg T., Am. J. Hum. Genet. 1999, 65, 664; - PMC - PubMed
- c) Sasaguri H., Nilsson P., Hashimoto S., Nagata K., Saito T., De Strooper B., Hardy J., Vassar R., Winblad B., Saido T. C., EMBO J. 2017, 36, 2473; - PMC - PubMed
- d) Long J. M., Holtzman D. M., Cell 2019, 179, 312. - PMC - PubMed
-
- a) Zhang L., Chen C., Mak M. S., Lu J., Wu Z., Chen Q., Han Y., Li Y., Pi R., Med. Res. Rev. 2020, 40, 431; - PubMed
- b) Drummond E., Wisniewski T., Acta Neuropathol. 2017, 133, 155; - PMC - PubMed
- c) Yiannopoulou K. G., Anastasiou A. I., Zachariou V., Pelidou S. H., Biomedicines 2019, 7, 97. - PMC - PubMed
-
- a) Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S., Cell 2007, 131, 861. - PubMed
- b) Yu J., Vodyanik M. A., Smuga‐Otto K., Antosiewicz‐Bourget J., Frane J. L., Tian S., Nie J., Jonsdottir G. A., Ruotti V., Stewart R., Slukvin I. I., Thomson J. A., Science 2007, 318, 1917. - PubMed
-
- a) Shi Y., Inoue H., Wu J. C., Yamanaka S., Nat. Rev. Drug Discovery 2017, 16, 115; - PMC - PubMed
- b) Rowe R. G., Daley G. Q., Nat. Rev. Genet. 2019, 20, 377; - PMC - PubMed
- c) Li L., Tian E., Chen X., Chao J., Klein J., Qu Q., Sun G., Sun G., Huang Y., Warden C. D., Ye P., Feng L., Li X., Cui Q., Sultan A., Douvaras P., Fossati V., Sanjana N. E., Riggs A. D., Shi Y., Cell Stem Cell 2018, 23, 239. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- Sidell Kagan Foundation
- R56 AG061171/AG/NIA NIH HHS/United States
- R01 AG056303/AG/NIA NIH HHS/United States
- P30 AG066519/AG/NIA NIH HHS/United States
- Christopher Family Endowed Innovation Fund
- RF1 AG061794/AG/NIA NIH HHS/United States
- R01 AG056305/AG/NIA NIH HHS/United States
- RF1 DA048813/DA/NIDA NIH HHS/United States
- P30 CA033572/CA/NCI NIH HHS/United States
- U24 NS072026/NS/NINDS NIH HHS/United States
- P30CA33572/National Cancer Institute of the National Institutes of Health
- P30 AG019610/AG/NIA NIH HHS/United States
- Louise and Herbert Horvitz Charitable Foundation
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous