

Defining Dravet syndrome: An essential pre-requisite for precision medicine trials
- PMID: 34338318
- PMCID: PMC9291974
- DOI: 10.1111/epi.17015
Defining Dravet syndrome: An essential pre-requisite for precision medicine trials
Abstract
Objective: The classical description of Dravet syndrome, the prototypic developmental and epileptic encephalopathy, is of a normal 6-month-old infant presenting with a prolonged, febrile, hemiclonic seizure and showing developmental slowing after age 1 year. SCN1A pathogenic variants are found in >80% of patients. Many patients have atypical features resulting in diagnostic delay and inappropriate therapy. We aimed to provide an evidence-based definition of SCN1A-Dravet syndrome in readiness for precision medicine trials.
Methods: Epilepsy patients were recruited to the University of Melbourne Epilepsy Genetics Research Program between 1995 and 2020 by neurologists from around the world. Patients with SCN1A pathogenic variants were reviewed and only those with Dravet syndrome were included. Clinical data, including seizure and developmental course, were analyzed in all patients with SCN1A-Dravet syndrome.
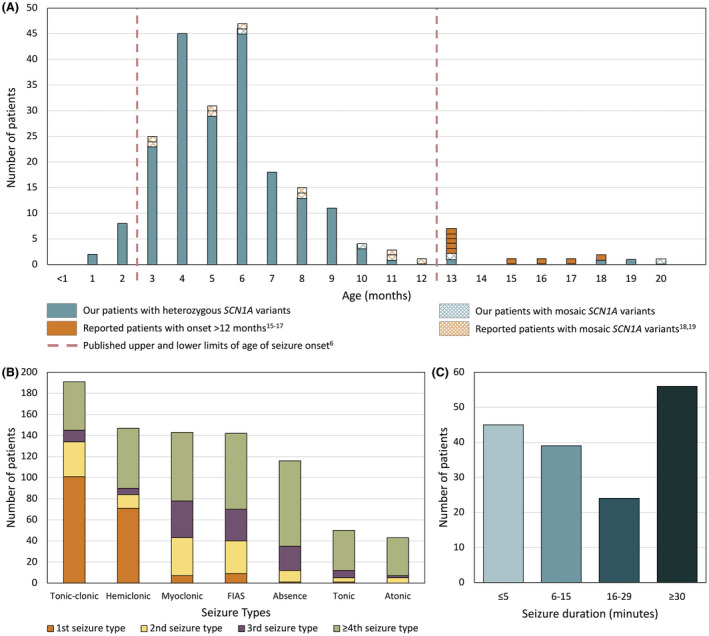
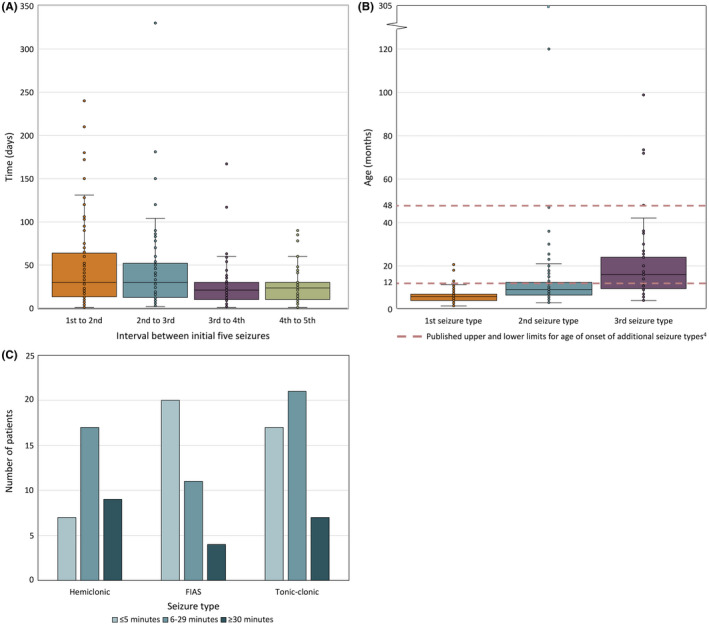
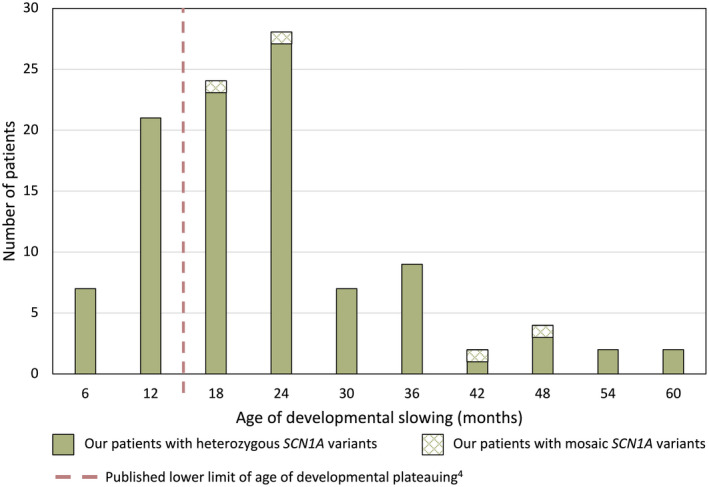
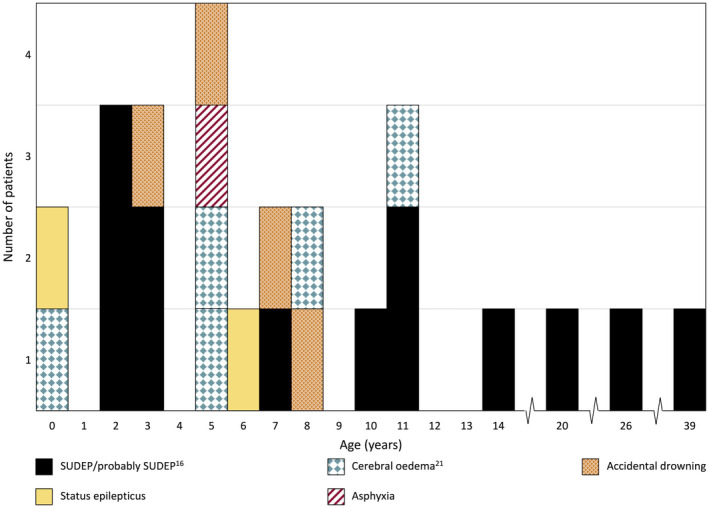
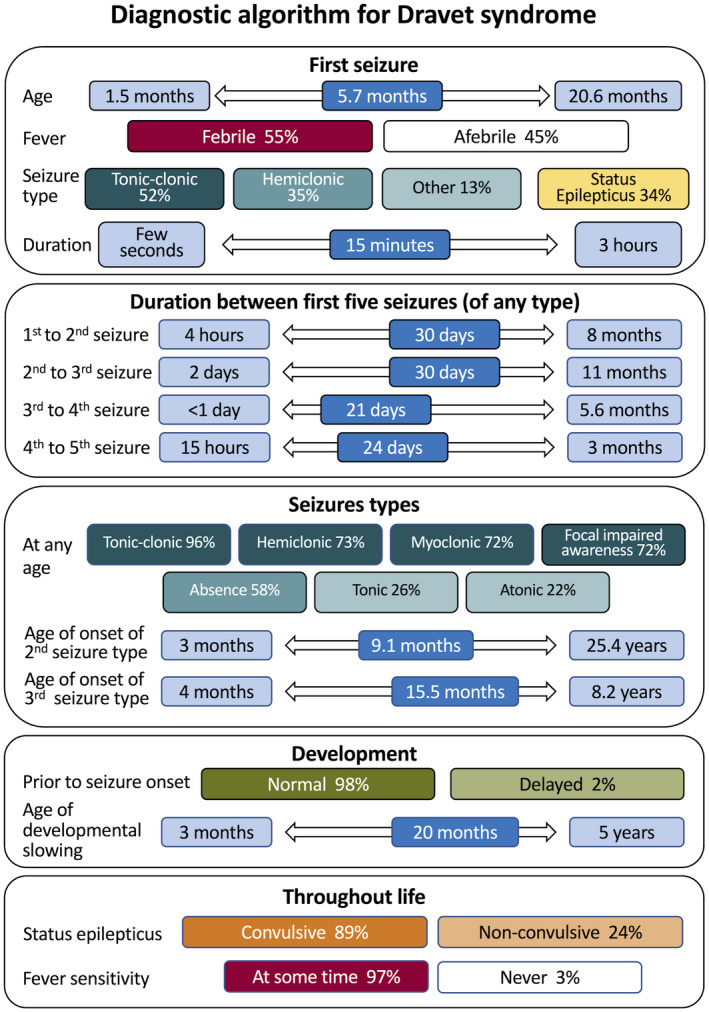
Results: Two hundred and five patients were studied at a median age of 8.5 years (range 10 months to 60 years); 25 were deceased. The median seizure-onset age was 5.7 months (range 1.5-20.6 months). Initial seizures were tonic-clonic (52%) and hemiclonic (35%), with only 55% being associated with fever. Only 34% of patients presented with status epilepticus (seizure lasting ≥30 minutes). Median time between first and second seizure was 30 days (range 4 hours to 8 months), and seven patients (5%) had at least 6 months between initial seizures. Median ages at onset of second and third seizure types were 9.1 months (range 3 months-25.4 years) and 15.5 months (range 4 months-8.2 years), respectively. Developmental slowing occurred prior to 12 months in 27%.
Significance: An evidence-based definition of SCN1A-Dravet syndrome is essential for early diagnosis. We refine the spectrum of Dravet syndrome, based on patterns of seizure onset, type, and progression. Understanding of the full spectrum of SCN1A-Dravet syndrome presentation is essential for early diagnosis and optimization of treatment, especially as precision medicine trials become available.
Keywords: SCN1A; Dravet syndrome; developmental and epileptic encephalopathy; genetics; phenotypic spectrum.
© 2021 The Authors. Epilepsia published by Wiley Periodicals LLC on behalf of International League Against Epilepsy.
Conflict of interest statement
I.E. Scheffer has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, Chiesi, Encoded Therapeutics, Xenon Pharmaceuticals, and Knopp Biosciences; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, Biomarin, and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx, GW Pharma, UCB, Eisai, Anavex Life Sciences, Ovid Therapeutics, Epigenyx, Encoded Therapeutics and Marinus; and has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics, Care Beyond Diagnosis, Epilepsy Consortium, and UCB. She may accrue future revenue on a pending patent WO2009/086591 (filed: 2008); has a patent for
Figures
References
-
- McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–16. - PubMed
-
- Dravet C, Bureau M, Oguni H, Cokar O, Guerrini R, Wolf P. Dravet syndrome (previously severe myoclonic epilepsy in infancy). In: Bureau MPG, Dravet C, Delgado‐Escueta AV, Guerrini R, Tassinari CA, Thomas P, eds. Epileptic Syndromes in Infancy, Childhood and Adolescence, 6th edn. Arcueil, France: John Libbey Eurotext; 2019: 139–171.
-
- Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;26:12. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
