

BDNF controls GABAAR trafficking and related cognitive processes via autophagic regulation of p62
- PMID: 34341497
- PMCID: PMC8674239
- DOI: 10.1038/s41386-021-01116-0
BDNF controls GABAAR trafficking and related cognitive processes via autophagic regulation of p62
Abstract
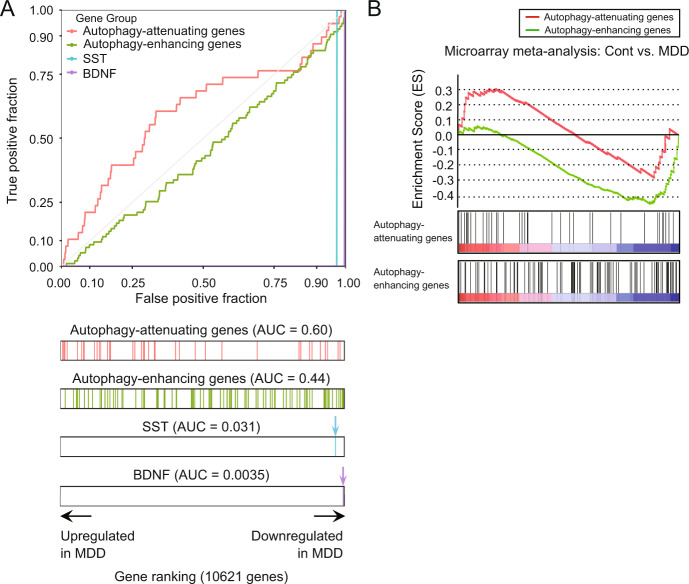
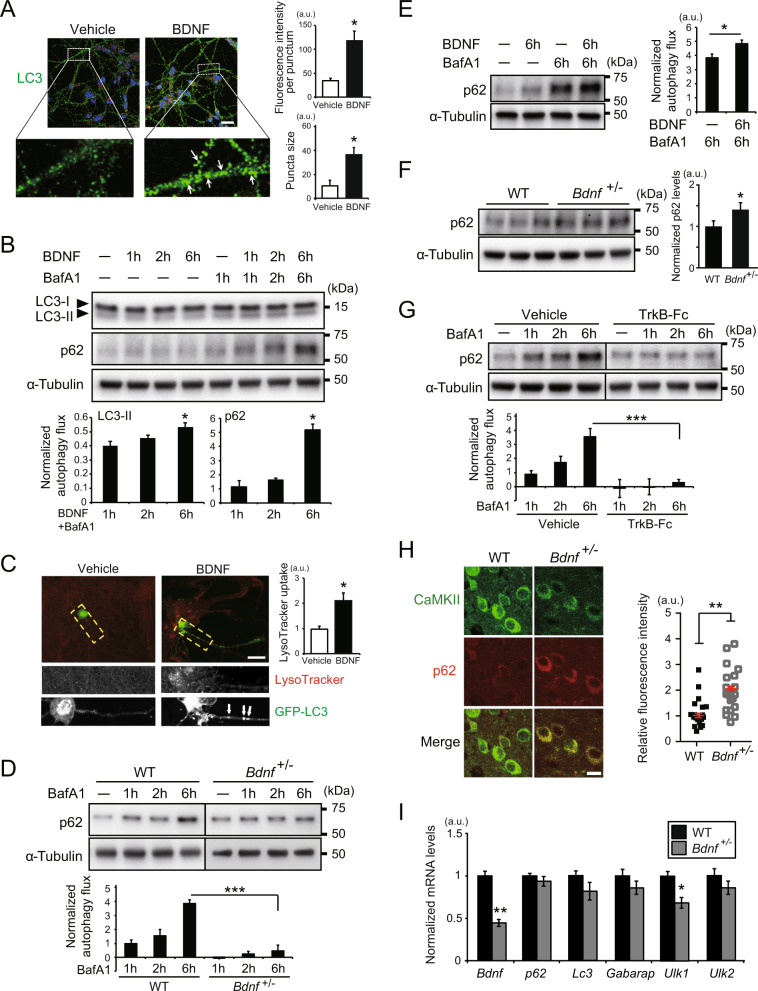
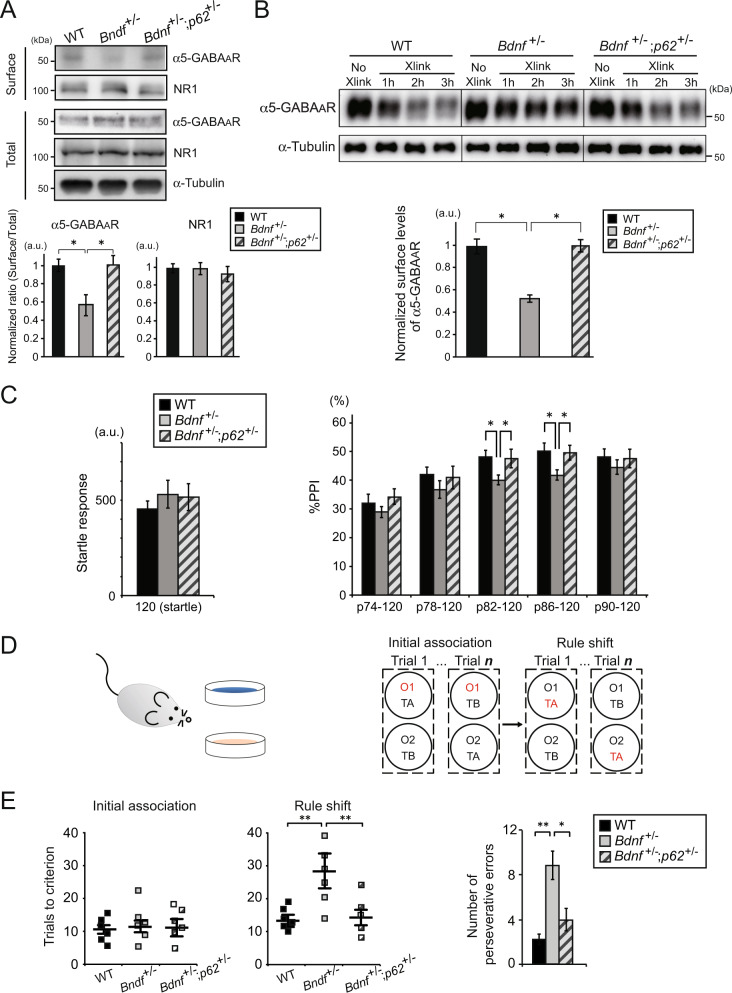
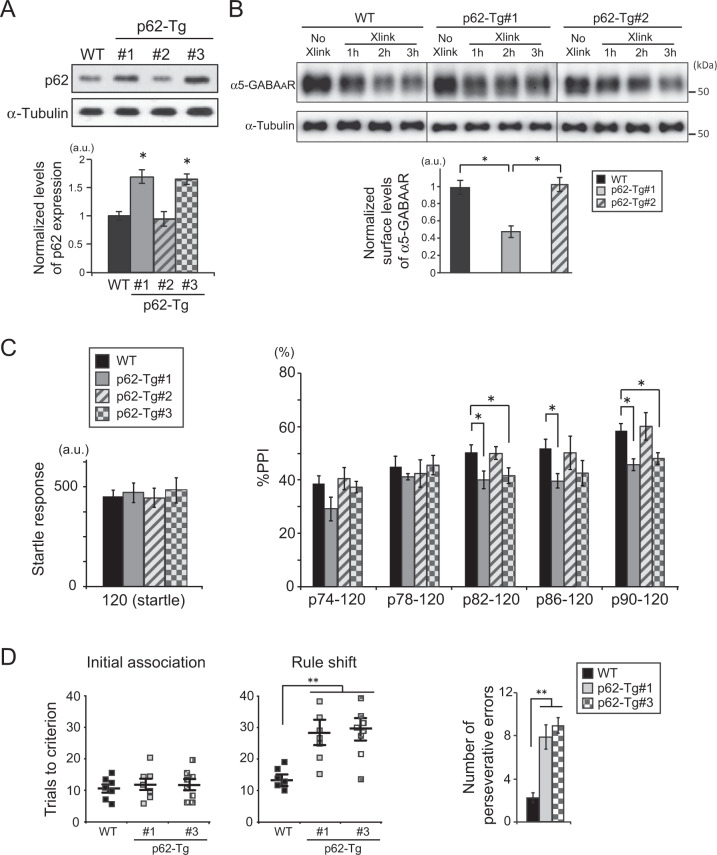
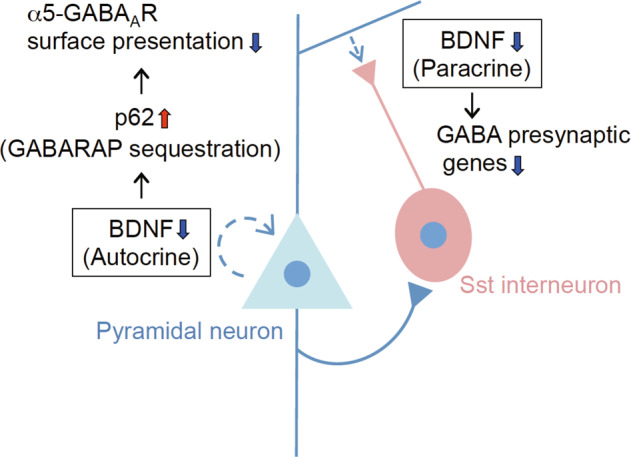
Reduced brain-derived neurotrophic factor (BDNF) and gamma-aminobutyric acid (GABA) neurotransmission co-occur in brain conditions (depression, schizophrenia and age-related disorders) and are associated with symptomatology. Rodent studies show they are causally linked, suggesting the presence of biological pathways mediating this link. Here we first show that reduced BDNF and GABA also co-occur with attenuated autophagy in human depression. Using mice, we then show that reducing Bdnf levels (Bdnf+/-) leads to upregulated sequestosome-1/p62, a key autophagy-associated adaptor protein, whose levels are inversely correlated with autophagic activity. Reduced Bdnf levels also caused reduced surface presentation of α5 subunit-containing GABAA receptor (α5-GABAAR) in prefrontal cortex (PFC) pyramidal neurons. Reducing p62 gene dosage restored α5-GABAAR surface expression and rescued PFC-relevant behavioral deficits of Bdnf+/- mice, including cognitive inflexibility and reduced sensorimotor gating. Increasing p62 levels was sufficient to recreate the molecular and behavioral profiles of Bdnf+/- mice. Collectively, the data reveal a novel mechanism by which deficient BDNF leads to targeted reduced GABAergic signaling through autophagic dysregulation of p62, potentially underlying cognitive impairment across brain conditions.
© 2021. The Author(s), under exclusive licence to American College of Neuropsychopharmacology.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Ulk2 controls cortical excitatory-inhibitory balance via autophagic regulation of p62 and GABAA receptor trafficking in pyramidal neurons.Hum Mol Genet. 2018 Sep 15;27(18):3165-3176. doi: 10.1093/hmg/ddy219. Hum Mol Genet. 2018. PMID: 29893844 Free PMC article.
-
Depolarizing, inhibitory GABA type A receptor activity regulates GABAergic synapse plasticity via ERK and BDNF signaling.Neuropharmacology. 2018 Jan;128:324-339. doi: 10.1016/j.neuropharm.2017.10.022. Epub 2017 Oct 23. Neuropharmacology. 2018. PMID: 29074304 Free PMC article.
-
Pro-brain-derived neurotrophic factor inhibits GABAergic neurotransmission by activating endocytosis and repression of GABAA receptors.J Neurosci. 2014 Oct 1;34(40):13516-34. doi: 10.1523/JNEUROSCI.2069-14.2014. J Neurosci. 2014. PMID: 25274828 Free PMC article.
-
GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon.Psychopharmacology (Berl). 2005 Jul;180(2):191-205. doi: 10.1007/s00213-005-2212-8. Epub 2005 Apr 28. Psychopharmacology (Berl). 2005. PMID: 15864560 Review.
-
Interaction of different proteins with GABAA receptor and their modulatory effect on inhibitory neural transmission leads to epilepsy.CNS Neurol Disord Drug Targets. 2014;13(7):1148-59. doi: 10.2174/1871527313666140917115121. CNS Neurol Disord Drug Targets. 2014. PMID: 25230226 Review.
Cited by
-
The cAMP Response Element- Binding Protein/Brain-Derived Neurotrophic Factor Pathway in Anterior Cingulate Cortex Regulates Neuropathic Pain and Anxiodepression Like Behaviors in Rats.Front Mol Neurosci. 2022 Mar 24;15:831151. doi: 10.3389/fnmol.2022.831151. eCollection 2022. Front Mol Neurosci. 2022. PMID: 35401106 Free PMC article.
-
Aberrant aging-associated p62 autophagic cascade in biopsied olfactory neuronal cells from patients with psychosis.Schizophrenia (Heidelb). 2025 Apr 23;11(1):68. doi: 10.1038/s41537-025-00617-x. Schizophrenia (Heidelb). 2025. PMID: 40268926 Free PMC article.
-
Reversal of Age-Related Neuronal Atrophy by α5-GABAA Receptor Positive Allosteric Modulation.Cereb Cortex. 2021 Jan 5;31(2):1395-1408. doi: 10.1093/cercor/bhaa310. Cereb Cortex. 2021. PMID: 33068001 Free PMC article.
-
Neuroinflammation alters GABAergic neurotransmission in hyperammonemia and hepatic encephalopathy, leading to motor incoordination. Mechanisms and therapeutic implications.Front Pharmacol. 2024 Mar 15;15:1358323. doi: 10.3389/fphar.2024.1358323. eCollection 2024. Front Pharmacol. 2024. PMID: 38560359 Free PMC article. Review.
-
Extrasynaptic localization is essential for α5GABAA receptor modulation of dopamine system function.eNeuro. 2024 Feb 27;11(3):ENEURO.0344-23.2023. doi: 10.1523/ENEURO.0344-23.2023. Online ahead of print. eNeuro. 2024. PMID: 38413199 Free PMC article.
References
-
- Castrén E, Kojima M. Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiol Dis. 2017;97:119–26. - PubMed
-
- Knable MB, Barci BM, Webster MJ, Meador-Woodruff J, Torrey EF, Stanley Neuropathology Consortium Molecular abnormalities of the hippocampus in severe psychiatric illness: postmortem findings from the Stanley Neuropathology Consortium. Mol Psychiatry. 2004;9:609–20. - PubMed
-
- Dunham JS, Deakin JFW, Miyajima F, Payton A, Toro CT. Expression of hippocampal brain-derived neurotrophic factor and its receptors in Stanley consortium brains. J Psychiatr Res. 2009;43:1175–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
