

Canine tumor mutational burden is correlated with TP53 mutation across tumor types and breeds
- PMID: 34344882
- PMCID: PMC8333103
- DOI: 10.1038/s41467-021-24836-9
Canine tumor mutational burden is correlated with TP53 mutation across tumor types and breeds
Abstract
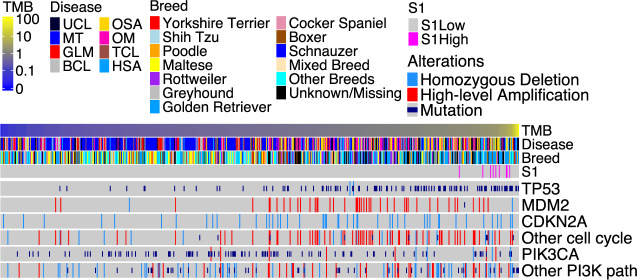
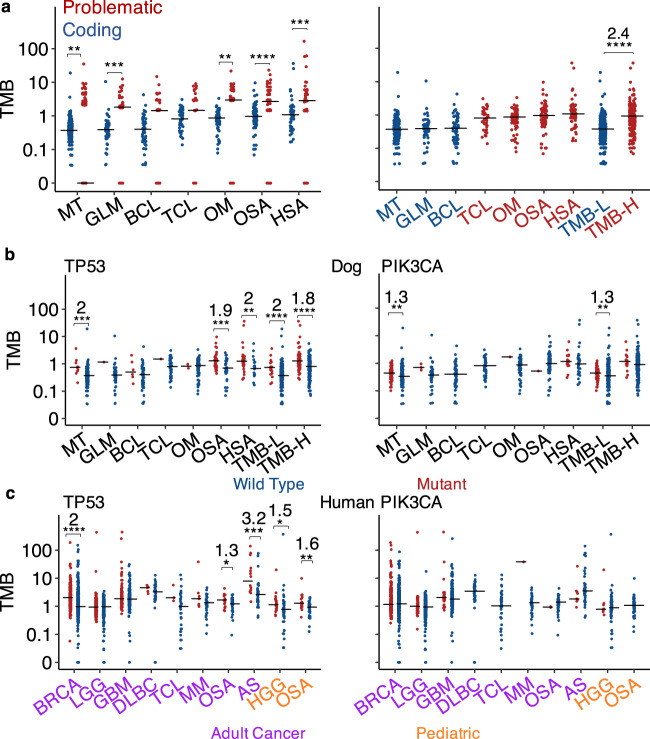
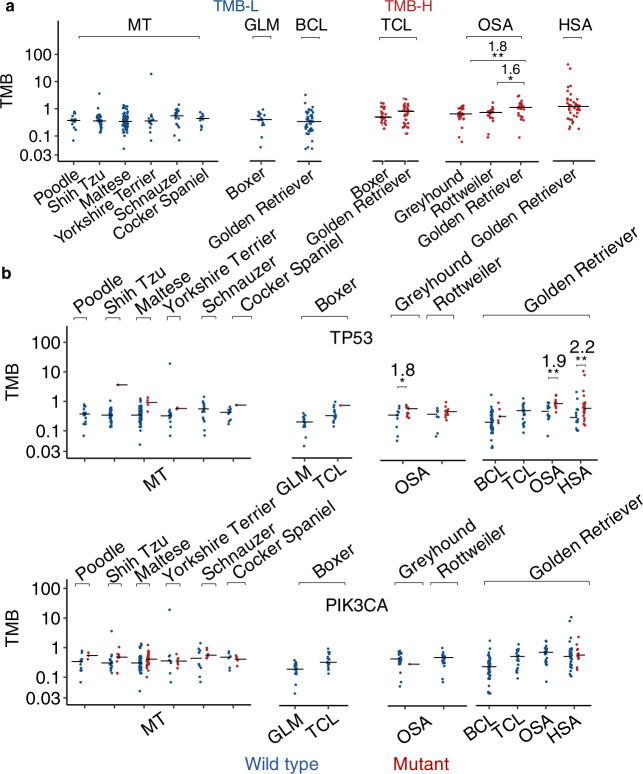
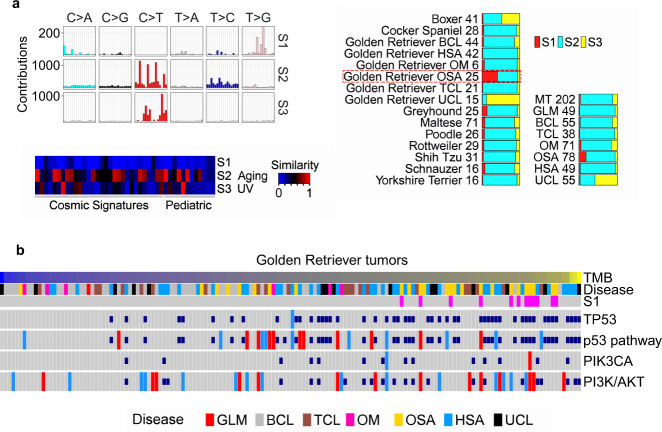
Spontaneous canine cancers are valuable but relatively understudied and underutilized models. To enhance their usage, we reanalyze whole exome and genome sequencing data published for 684 cases of >7 common tumor types and >35 breeds, with rigorous quality control and breed validation. Our results indicate that canine tumor alteration landscape is tumor type-dependent, but likely breed-independent. Each tumor type harbors major pathway alterations also found in its human counterpart (e.g., PI3K in mammary tumor and p53 in osteosarcoma). Mammary tumor and glioma have lower tumor mutational burden (TMB) (median < 0.5 mutations per Mb), whereas oral melanoma, osteosarcoma and hemangiosarcoma have higher TMB (median ≥ 1 mutations per Mb). Across tumor types and breeds, TMB is associated with mutation of TP53 but not PIK3CA, the most mutated genes. Golden Retrievers harbor a TMB-associated and osteosarcoma-enriched mutation signature. Here, we provide a snapshot of canine mutations across major tumor types and breeds.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
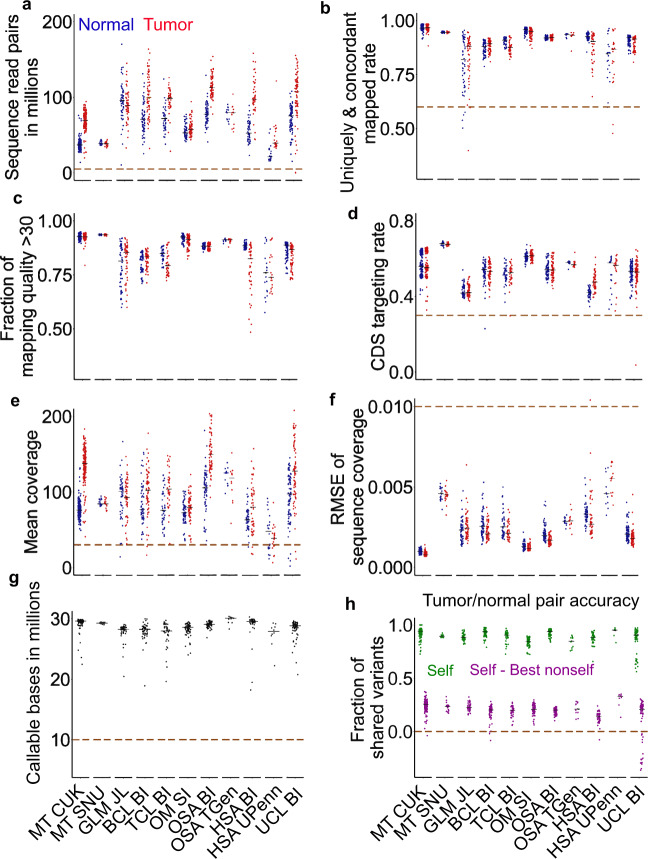
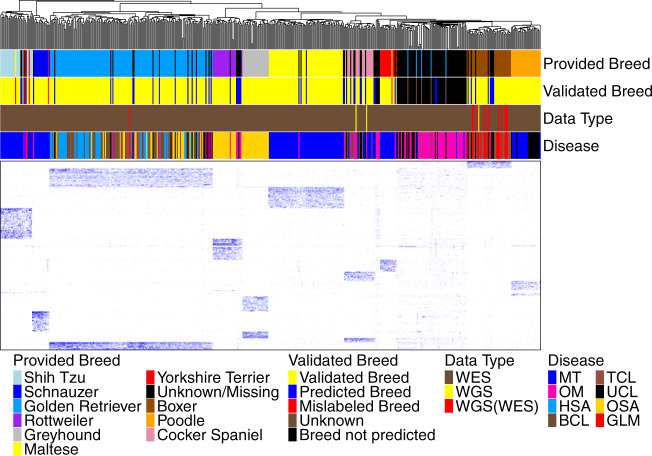
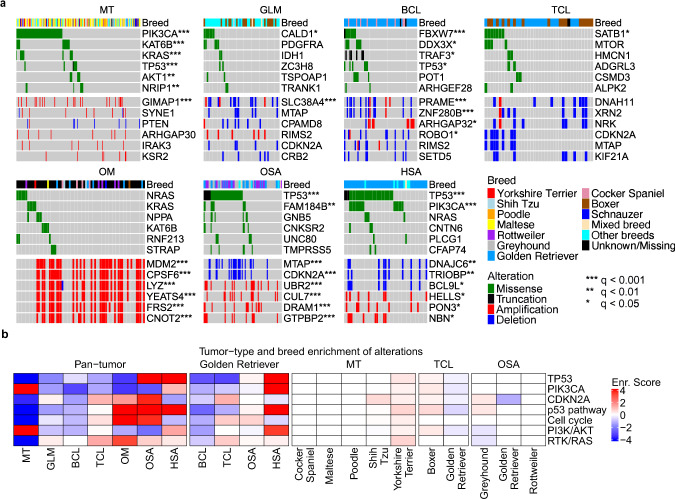
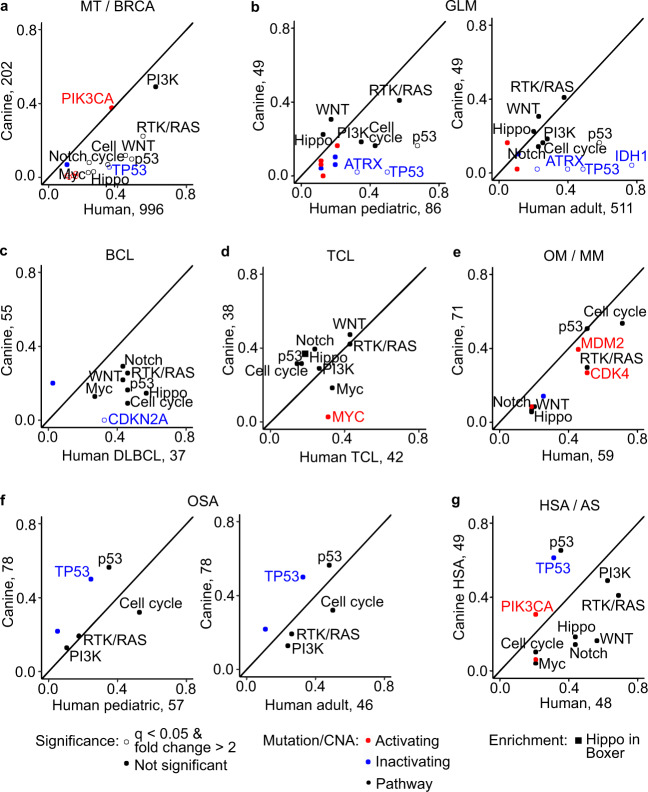
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
