

doi: 10.1056/NEJMsb2104756.
SARS-CoV-2 Variants in Patients with Immunosuppression
Affiliations
- PMID: 34347959
- PMCID: PMC8494465
- DOI: 10.1056/NEJMsb2104756
Item in Clipboard
SARS-CoV-2 Variants in Patients with Immunosuppression
N Engl J Med.
.
No abstract available
Figures
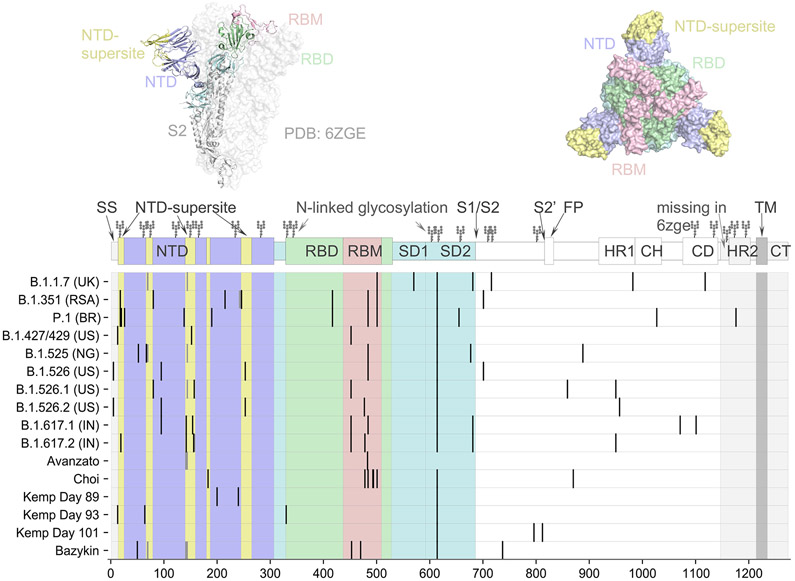
Panel A shows the spike protein structure of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (entry 6ZGE in the Worldwide Protein Data Bank [PDB]). Domains are colored according to their role in viral entry and antibody recognition; the terminal domain that is missing in 6ZGE is shaded in gray. As shown in the graphic above the chart, the spike protein consists of two subunits, with subunit 1 (S1) (the portions in yellow, purple, blue, green, and pink) containing the receptor-binding domain (RBD), the N-terminal domain (NTD), and several other subdomains and subunit 2 (S2) (the portion in gray) mediating fusion of the virus-cell membrane. Also shown are the spike mutations that are representative of circulating variants of concern (B.1.1.7, B.1.351, P.1, and B.1.617.2) or variants of interest (B.1.427/429, B.1.525, and B.1.526), as well as patterns of mutations identified over the course of persistent infections in immunocompromised patients, as described in case reports. Representative sequences from the case reports are listed according to the first author’s name (,,,,). Mutations in the NTD super- site are located nearby on the protein structure. Mutations are also recurring in the receptor-binding motif (RBM) and around the furin cleavage site. The symbols at the top of the columns indicate the N-linked glycosylation sites. CD denotes connector domain, CH center helix, CT cytoplasmic tail, HR1 heptad repeat 1, HR2 heptad repeat 2, SD1 subdomain 1, SD2 subdomain 2, S2′FP S2 fusion peptide, and TM transmembrane. Panel B shows the phylogenetic tree of 1510 SARS-CoV-2 viruses detected in patients in the United Kingdom between January 2020 and February 2021. The phylogeny is embedded as a root-to-tip plot, in which the x axis represents the date of sample collection and the y axis represents the number of genomewide mutations that have occurred since the phylogeny root. The phylogeny is colored according to the clade, as listed in the Nextstrain database (an online site for the sharing of sequencing and genomic data about SARS-CoV-2). Clade 20I/501Y.V1 in this database corresponds to lineage B.1.1.7 in the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGO) and is colored in orange and annotated in the figure. The branch leading to lineage B.1.1.7 is annotated with selected mutations that are present along this single branch. Lineage B.1.1.7 has a higher number of mutations than most other circulating viruses and has rapidly displaced existing genetic diversity in the United Kingdom. The phylogeny is constructed from sequence data shared to GISAID (a database of genomic information regarding influenza viruses and coronaviruses) by means of methods implemented by Nextstrain.
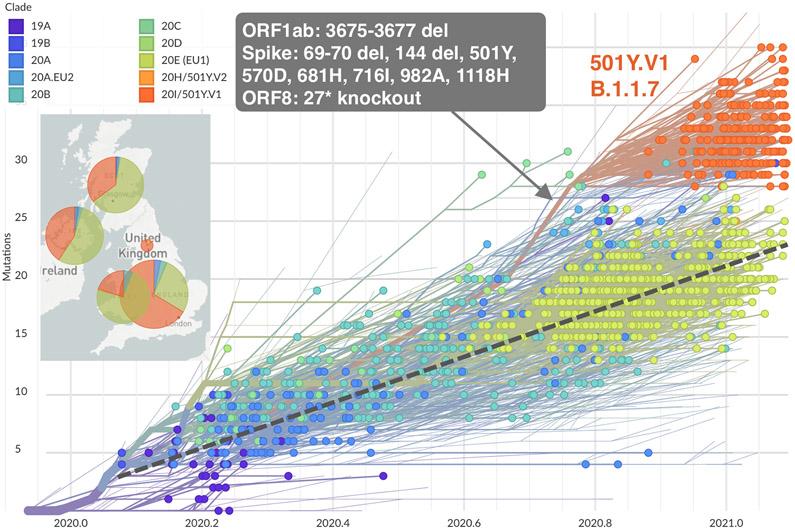
Panel A shows the spike protein structure of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (entry 6ZGE in the Worldwide Protein Data Bank [PDB]). Domains are colored according to their role in viral entry and antibody recognition; the terminal domain that is missing in 6ZGE is shaded in gray. As shown in the graphic above the chart, the spike protein consists of two subunits, with subunit 1 (S1) (the portions in yellow, purple, blue, green, and pink) containing the receptor-binding domain (RBD), the N-terminal domain (NTD), and several other subdomains and subunit 2 (S2) (the portion in gray) mediating fusion of the virus-cell membrane. Also shown are the spike mutations that are representative of circulating variants of concern (B.1.1.7, B.1.351, P.1, and B.1.617.2) or variants of interest (B.1.427/429, B.1.525, and B.1.526), as well as patterns of mutations identified over the course of persistent infections in immunocompromised patients, as described in case reports. Representative sequences from the case reports are listed according to the first author’s name (,,,,). Mutations in the NTD super- site are located nearby on the protein structure. Mutations are also recurring in the receptor-binding motif (RBM) and around the furin cleavage site. The symbols at the top of the columns indicate the N-linked glycosylation sites. CD denotes connector domain, CH center helix, CT cytoplasmic tail, HR1 heptad repeat 1, HR2 heptad repeat 2, SD1 subdomain 1, SD2 subdomain 2, S2′FP S2 fusion peptide, and TM transmembrane. Panel B shows the phylogenetic tree of 1510 SARS-CoV-2 viruses detected in patients in the United Kingdom between January 2020 and February 2021. The phylogeny is embedded as a root-to-tip plot, in which the x axis represents the date of sample collection and the y axis represents the number of genomewide mutations that have occurred since the phylogeny root. The phylogeny is colored according to the clade, as listed in the Nextstrain database (an online site for the sharing of sequencing and genomic data about SARS-CoV-2). Clade 20I/501Y.V1 in this database corresponds to lineage B.1.1.7 in the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGO) and is colored in orange and annotated in the figure. The branch leading to lineage B.1.1.7 is annotated with selected mutations that are present along this single branch. Lineage B.1.1.7 has a higher number of mutations than most other circulating viruses and has rapidly displaced existing genetic diversity in the United Kingdom. The phylogeny is constructed from sequence data shared to GISAID (a database of genomic information regarding influenza viruses and coronaviruses) by means of methods implemented by Nextstrain.
References
-
- Truong TT, Ryutov A, Pandey U, et al. Persistent SARS-CoV-2 infection and increasing viral variants in children and young adults with impaired humoral immunity. March 2, 2021. (https://www.medrxiv.org/content/10.1101/2021.02.27.21252099v1). preprint. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous