

Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets
- PMID: 34348167
- PMCID: PMC8803988
- DOI: 10.1016/j.preteyeres.2021.100998
Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets
Abstract
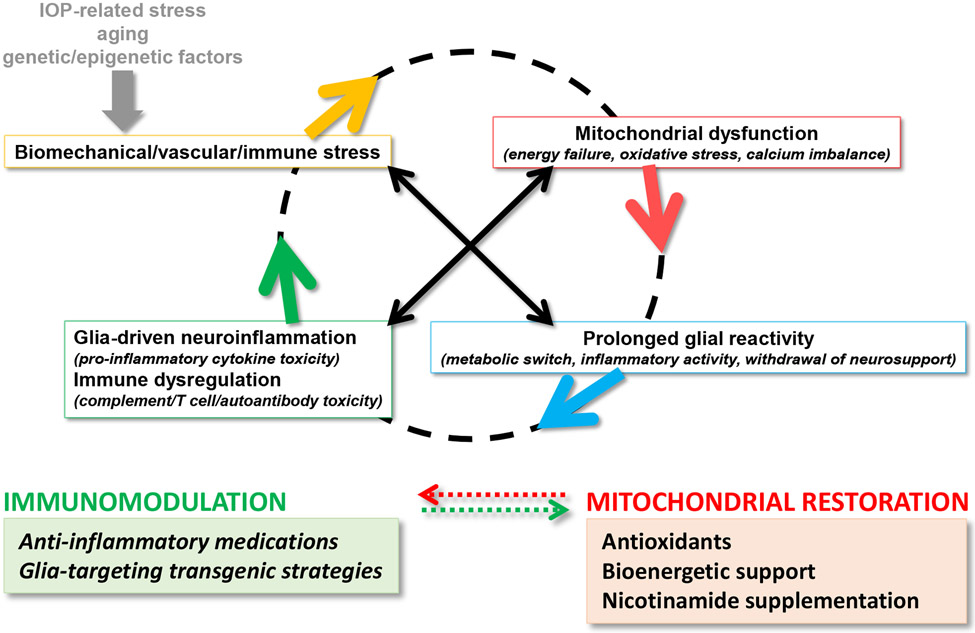
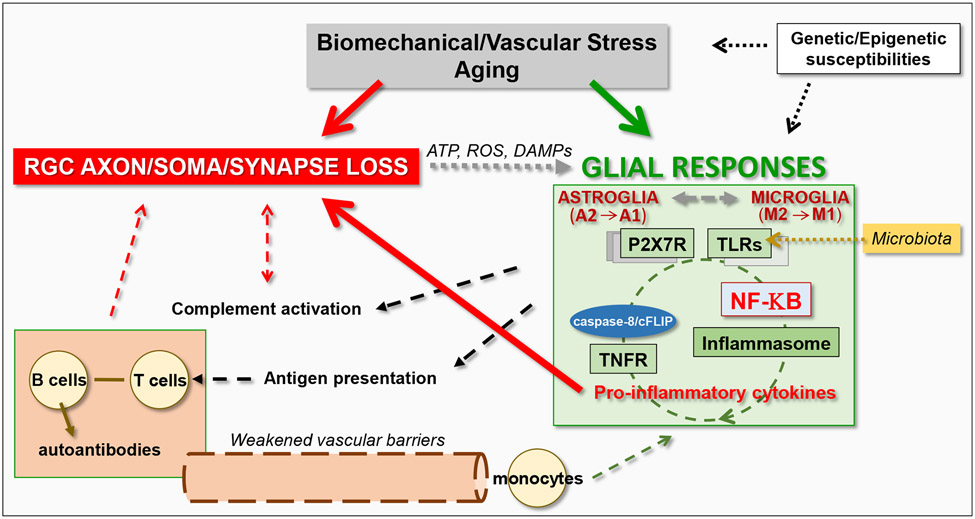
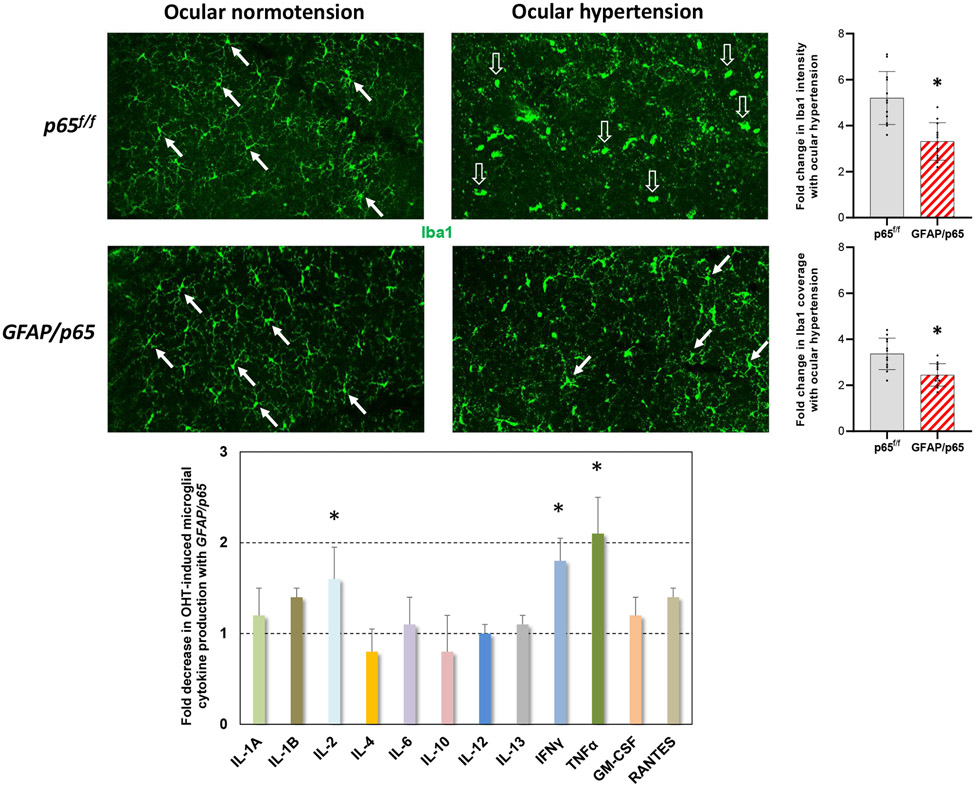
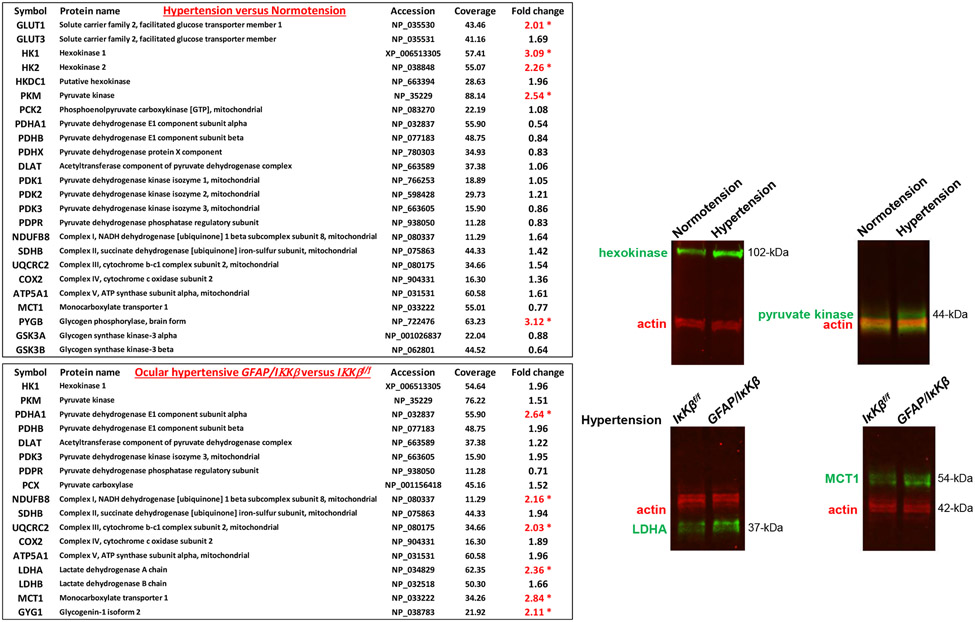
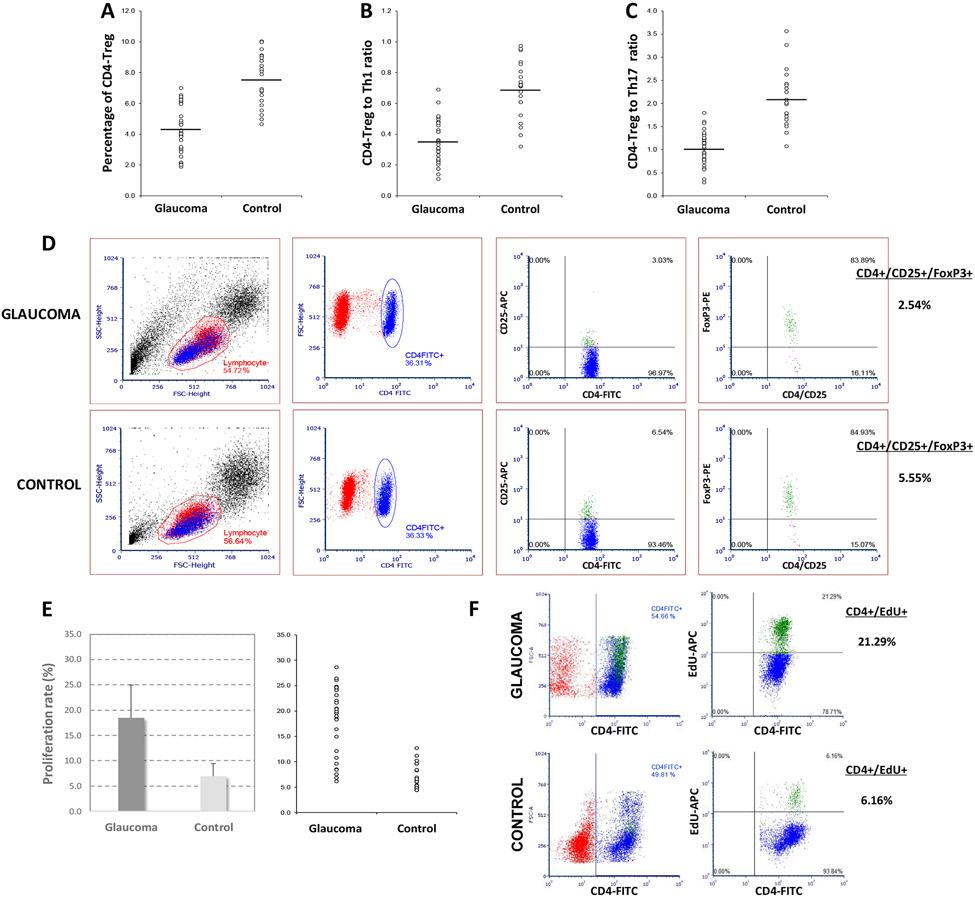
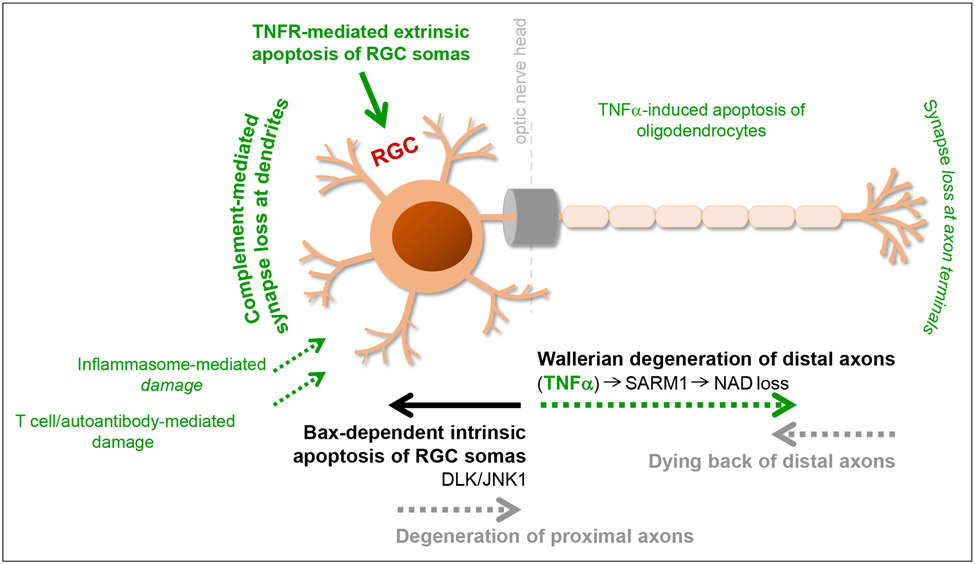
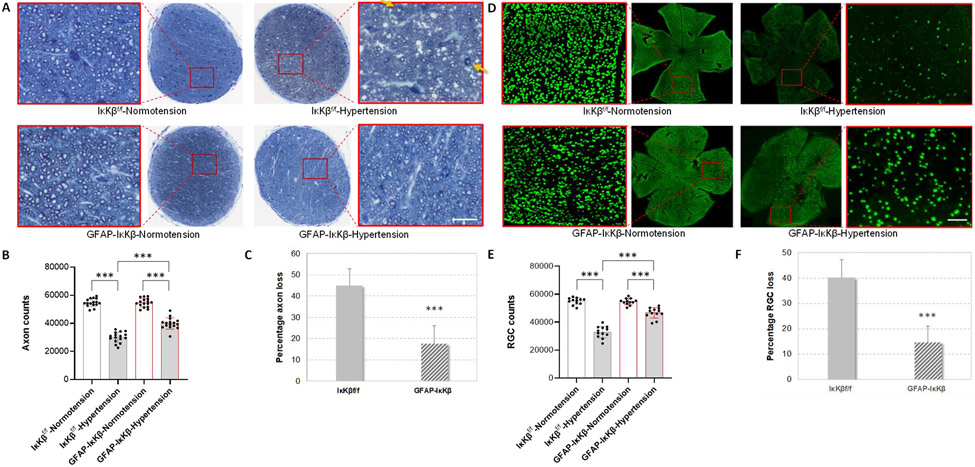
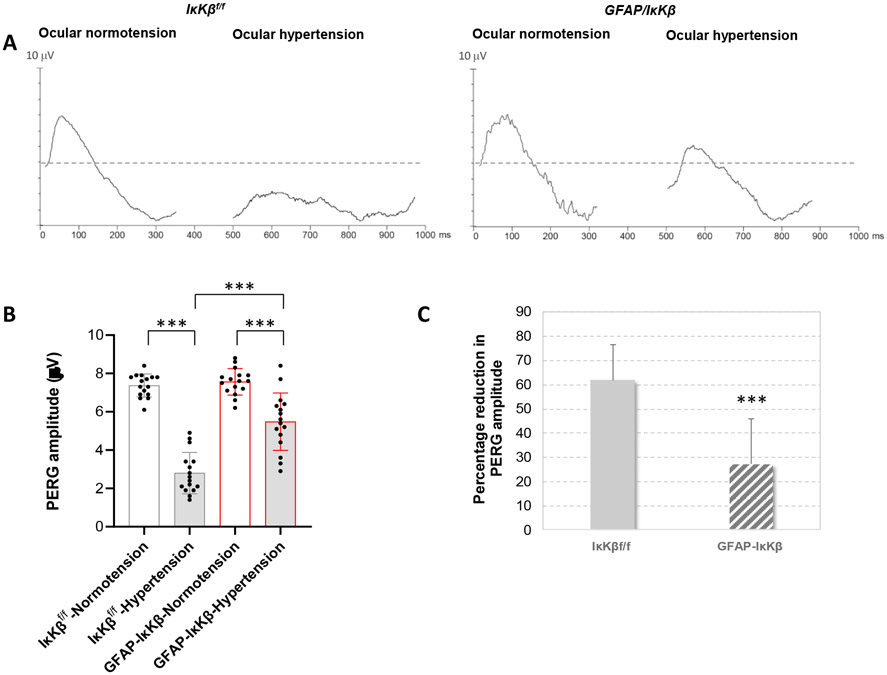
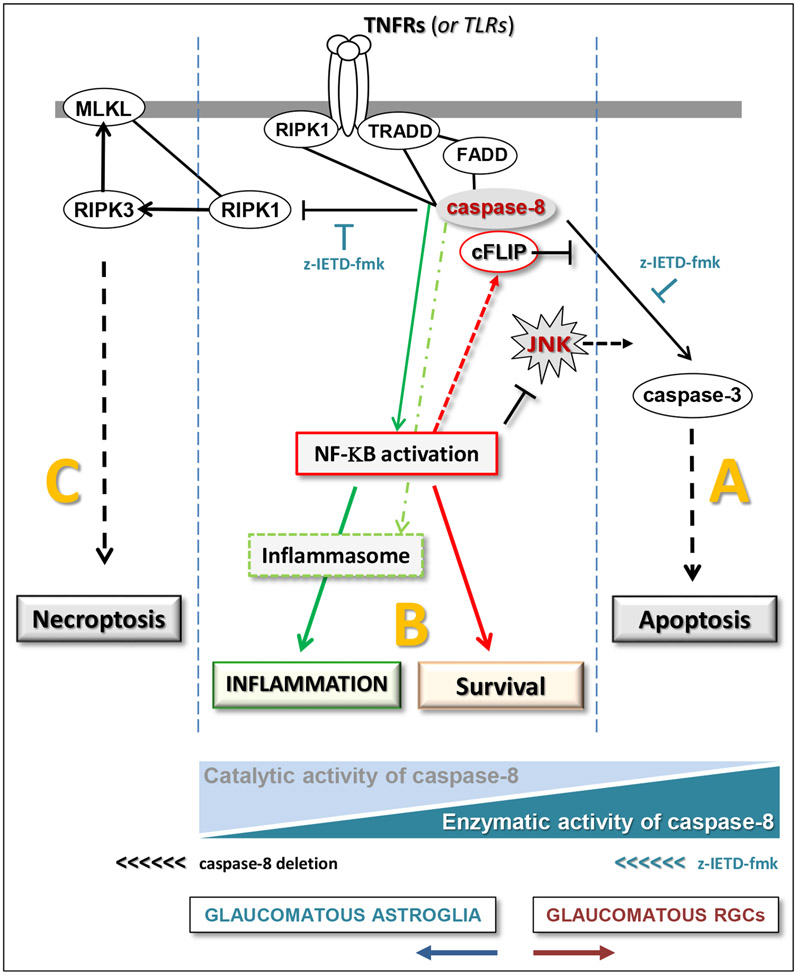
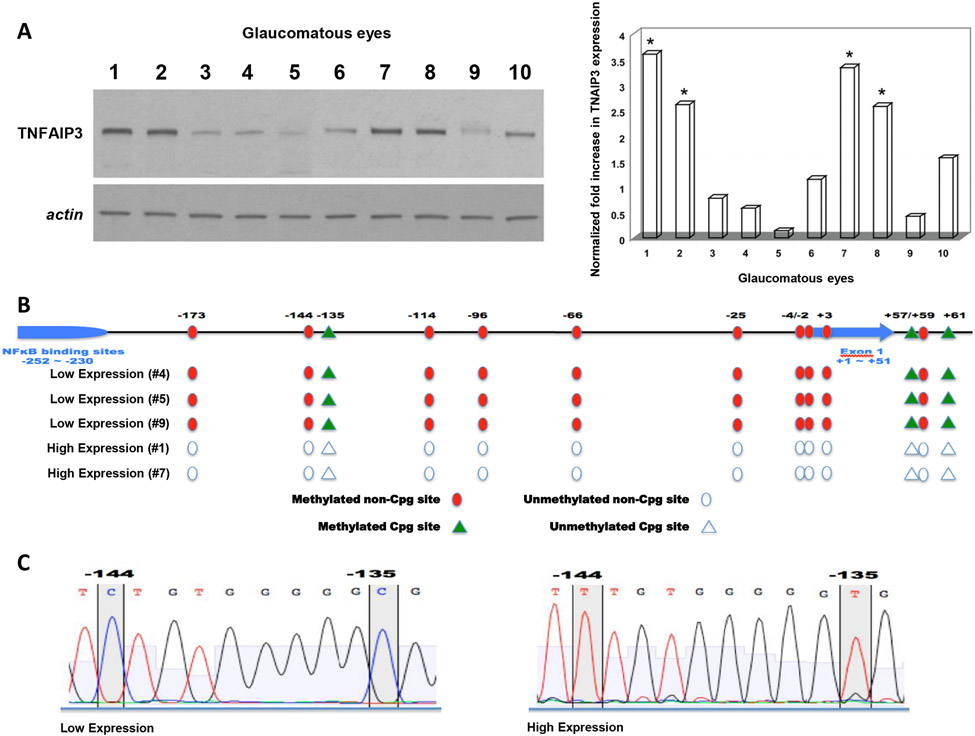
Neuroinflammation relying on the inflammatory responses of glial cells has emerged as an impactful component of the multifactorial etiology of neurodegeneration in glaucoma. It has become increasingly evident that despite early adaptive and reparative features of glial responses, prolonged reactivity of the resident glia, along with the peripheral immune cells, create widespread toxicity to retinal ganglion cell (RGC) axons, somas, and synapses. As much as the synchronized responses of astrocytes and microglia to glaucoma-related stress or neuron injury, their bi-directional interactions are critical to build and amplify neuroinflammation and to dictate the neurodegenerative outcome. Although distinct molecular programs regulate somatic and axonal degeneration in glaucoma, inhibition of neurodegenerative inflammation can provide a broadly beneficial treatment strategy to rescue RGC integrity and function. Since inflammatory toxicity and mitochondrial dysfunction are converging etiological paths that can boost each other and feed into a vicious cycle, anti-inflammatory treatments may also offer a multi-target potential. This review presents an overview of the current knowledge on neuroinflammation in glaucoma with particular emphasis on the cell-intrinsic and cell-extrinsic factors involved in the reciprocal regulation of glial responses, the interdependence between inflammatory and mitochondrial routes of neurodegeneration, and the research aspects inspiring for prospective immunomodulatory treatments. With the advent of powerful technologies, ongoing research on molecular and functional characteristics of glial responses is expected to accumulate more comprehensive and complementary information and to rapidly move the field forward to safe and effective modulation of the glial pro-inflammatory activities, while restoring or augmenting the glial immune-regulatory and neurosupport functions.
Keywords: Glaucoma; Glia; Immunomodulation; Neurodegeneration; Neuroinflammation; Retinal ganglion cell.
Copyright © 2021 The Author. Published by Elsevier Ltd.. All rights reserved.
Figures
References
-
- Aires ID, Ribeiro-Rodrigues T, Boia R, Catarino S, Girao H, Ambrosio AF, Santiago AR, 2020. Exosomes derived from microglia exposed to elevated pressure amplify the neuroinflammatory response in retinal cells. Glia. 68, 2705–2724. - PubMed
-
- Alarcon-Martinez L, Villafranca-Baughman D, Quintero H, et al., 2020. Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature. 585, 91–95. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
