

A novel SARS-CoV-2 related coronavirus with complex recombination isolated from bats in Yunnan province, China
- PMID: 34348599
- PMCID: PMC8381922
- DOI: 10.1080/22221751.2021.1964925
A novel SARS-CoV-2 related coronavirus with complex recombination isolated from bats in Yunnan province, China
Abstract
At the end of 2019, A new type of beta-CoV, SARS-CoV-2 emerged and triggered the COVID-19 pandemic, which spread overwhelmingly around the world in less than a year. However, the origin and direct ancestral viruses of SARS-CoV-2 remain unknown. RaTG13, a novel coronavirus found in bats in China's Yunnan Province, is the closest relative virus of the SARS-CoV-2 identified so far. In this study, a new SARS-CoV-2 related virus, provisionally named PrC31, was discovered in Yunnan province by retrospectively analyse bat next generation sequencing (NGS) data of intestinal samples collected in 2018. PrC31 shared 90.7% and 92.0% nucleotide identities to the genomes of SARS-CoV-2 and the bat SARSr-CoV ZC45, respectively. Sequence alignment of PrC31 showed that several genomic regions, especially orf1a and orf8 had the highest homology with those corresponding genomic regions of SARS-CoV-2 than any other related viruses. Phylogenetic analysis indicated that PrC31 shared a common ancestor with SARS-CoV-2 in evolutionary history. The differences between the PrC31 and SARS-CoV-2 genomes were mainly manifested in the spike genes. The amino acid homology between the receptor binding domains of PrC31 and SARS-CoV-2 was only 64.2%. Importantly, recombination analysis revealed that PrC31 underwent multiple complex recombination events (including three recombination breakpoints) involving the SARS-CoV and SARS-CoV-2 sub-lineages, indicating that PrC31 evolved from yet-to-be-identified intermediate recombination strains. Combined with previous studies, it is revealed that the beta-CoVs may possess a more complex recombination mechanism than we thought.
Keywords: SARS-CoV-2; bats; coronaviruses; evolution; recombination.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
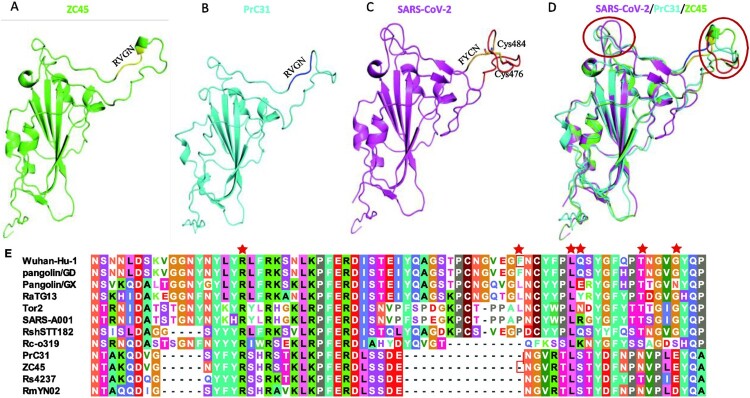
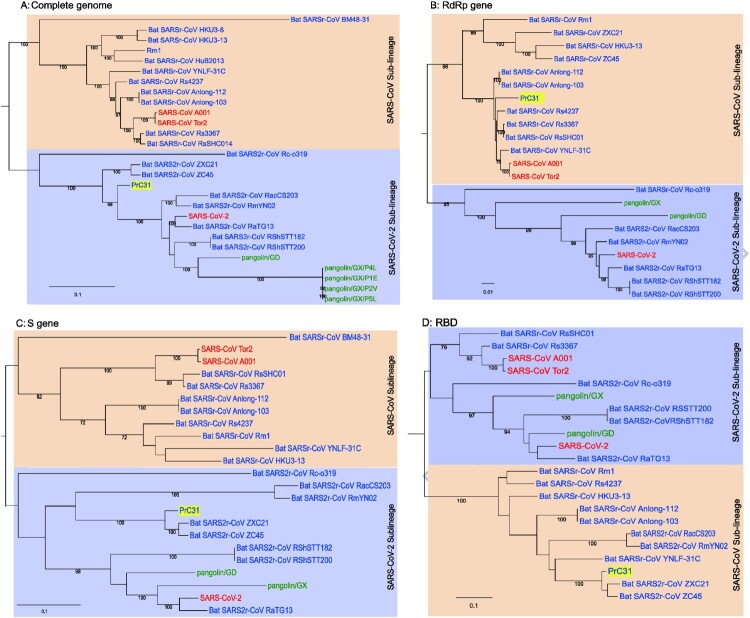
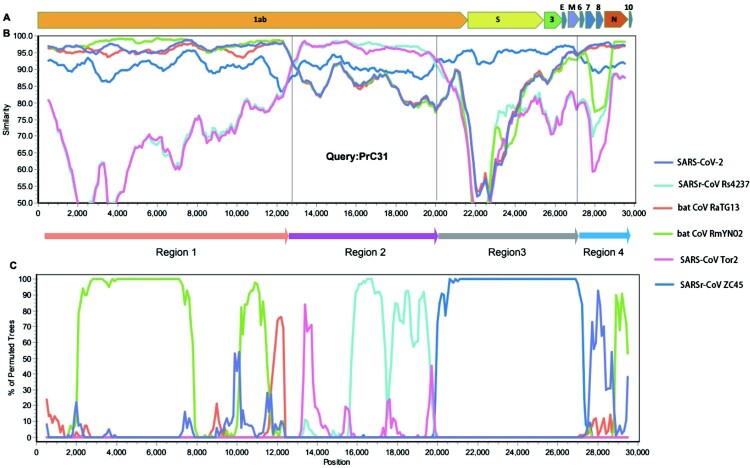
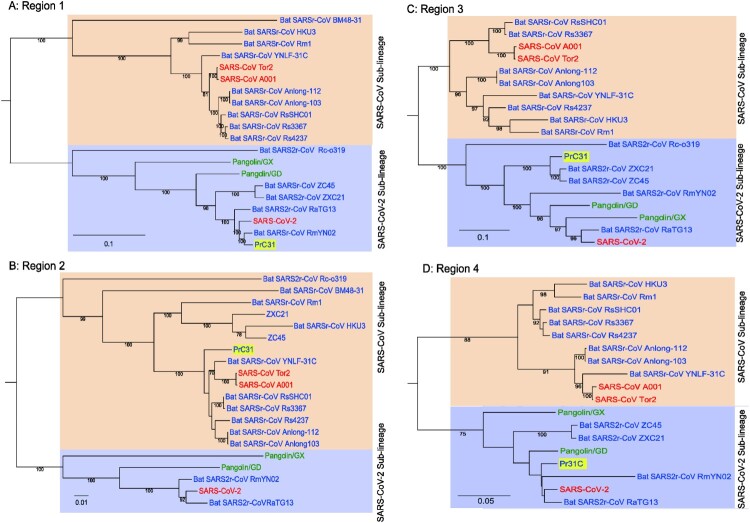
Similar articles
-
Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination.J Virol. 2015 Oct;89(20):10532-47. doi: 10.1128/JVI.01048-15. Epub 2015 Aug 12. J Virol. 2015. PMID: 26269185 Free PMC article.
-
Epidemiology and Genomic Characterization of Two Novel SARS-Related Coronaviruses in Horseshoe Bats from Guangdong, China.mBio. 2022 Jun 28;13(3):e0046322. doi: 10.1128/mbio.00463-22. Epub 2022 Apr 25. mBio. 2022. PMID: 35467426 Free PMC article.
-
Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus.PLoS Pathog. 2017 Nov 30;13(11):e1006698. doi: 10.1371/journal.ppat.1006698. eCollection 2017 Nov. PLoS Pathog. 2017. PMID: 29190287 Free PMC article.
-
Molecular epidemiology, evolution and phylogeny of SARS coronavirus.Infect Genet Evol. 2019 Jul;71:21-30. doi: 10.1016/j.meegid.2019.03.001. Epub 2019 Mar 4. Infect Genet Evol. 2019. PMID: 30844511 Free PMC article. Review.
-
[Source of the COVID-19 pandemic: ecology and genetics of coronaviruses (Betacoronavirus: Coronaviridae) SARS-CoV, SARS-CoV-2 (subgenus Sarbecovirus), and MERS-CoV (subgenus Merbecovirus).].Vopr Virusol. 2020;65(2):62-70. doi: 10.36233/0507-4088-2020-65-2-62-70. Vopr Virusol. 2020. PMID: 32515561 Review. Russian.
Cited by
-
Complete genome sequence of a novel bat mastadenovirus C strain isolated from Rhinolophus cornutus in Japan.Arch Virol. 2022 Mar;167(3):979-982. doi: 10.1007/s00705-021-05357-8. Epub 2022 Feb 3. Arch Virol. 2022. PMID: 35112204 Free PMC article.
-
Molecular diagnostic approaches for SARS-CoV-2 detection and pathophysiological consequences.Mol Biol Rep. 2023 Dec;50(12):10367-10382. doi: 10.1007/s11033-023-08844-0. Epub 2023 Oct 10. Mol Biol Rep. 2023. PMID: 37817022 Review.
-
The high diversity of SARS-CoV-2-related coronaviruses in pangolins alerts potential ecological risks.Zool Res. 2021 Nov 18;42(6):834-844. doi: 10.24272/j.issn.2095-8137.2021.334. Zool Res. 2021. PMID: 34766482 Free PMC article.
-
A novel bat coronavirus with a polybasic furin-like cleavage site.Virol Sin. 2023 Jun;38(3):344-350. doi: 10.1016/j.virs.2023.04.009. Epub 2023 May 2. Virol Sin. 2023. PMID: 37141989 Free PMC article.
-
Management of SARS-CoV-2 Infection-Clinical Practice Guidelines of the Polish Association of Epidemiologists and Infectiologists, for 2025.J Clin Med. 2025 Mar 27;14(7):2305. doi: 10.3390/jcm14072305. J Clin Med. 2025. PMID: 40217755 Free PMC article. Review.
References
-
- CDC . Severe Acute Respiratory Syndrome: Available online:https://www.cdc.gov/sars/about/fs-sars.html.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous