

10 years of CEMARA database in the AnDDI-Rares network: a unique resource facilitating research and epidemiology in developmental disorders in France
- PMID: 34348744
- PMCID: PMC8335940
- DOI: 10.1186/s13023-021-01957-4
10 years of CEMARA database in the AnDDI-Rares network: a unique resource facilitating research and epidemiology in developmental disorders in France
Abstract
Background: In France, the Ministry of Health has implemented a comprehensive program for rare diseases (RD) that includes an epidemiological program as well as the establishment of expert centers for the clinical care of patients with RD. Since 2007, most of these centers have entered the data for patients with developmental disorders into the CEMARA population-based registry, a national online data repository for all rare diseases. Through the CEMARA web portal, descriptive demographic data, clinical data, and the chronology of medical follow-up can be obtained for each center. We address the interest and ongoing challenges of this national data collection system 10 years after its implementation.
Methods: Since 2007, clinicians and researchers have reported the "minimum dataset (MDS)" for each patient presenting to their expert center. We retrospectively analyzed administrative data, demographic data, care organization and diagnoses.
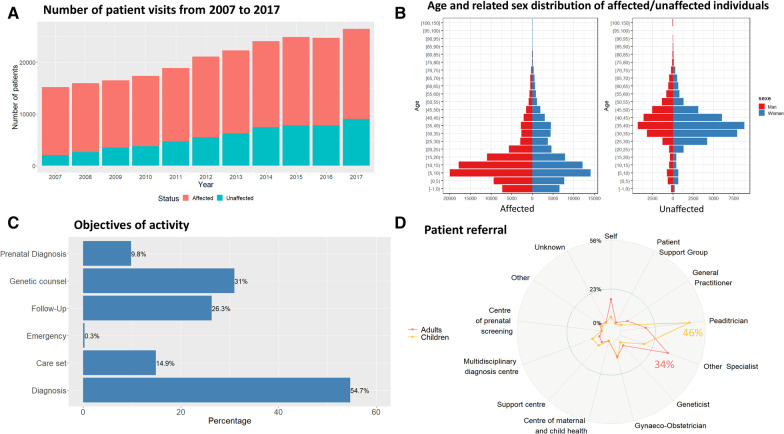
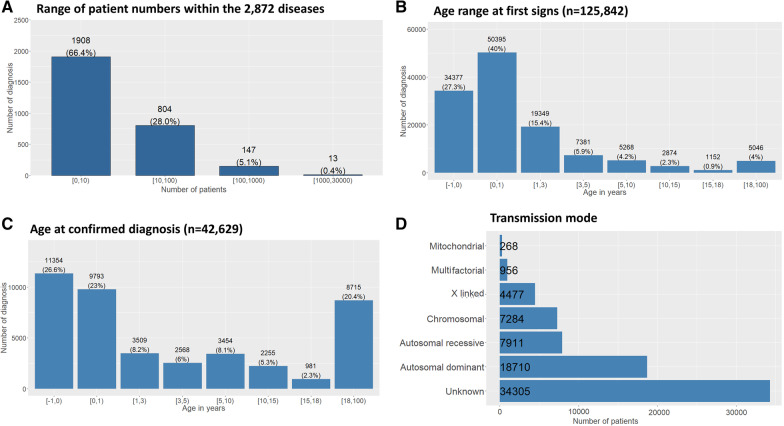
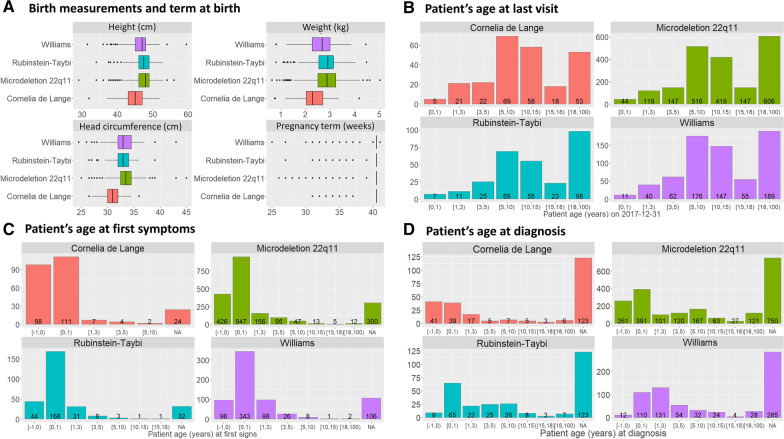
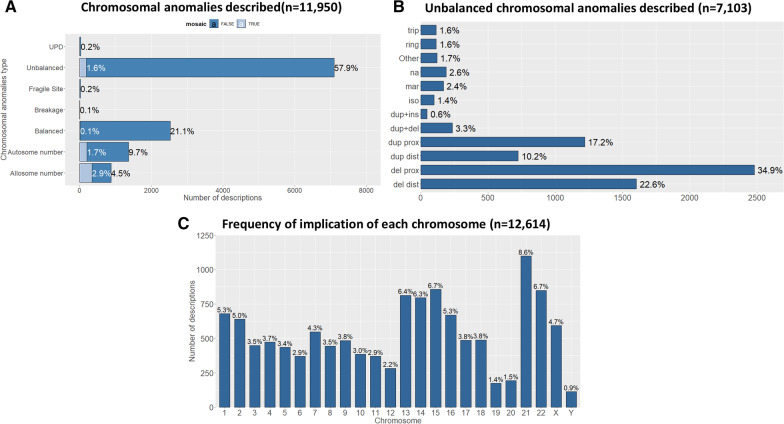
Results: Over 10 years, 228,243 RD patients (including healthy carriers and family members for whom experts denied any suspicion of RD) have visited an expert center. Among them, 167,361 were patients affected by a RD (median age 11 years, 54% children, 46% adults, with a balanced sex ratio), and 60,882 were unaffected relatives (median age 37 years). The majority of patients (87%) were seen no more than once a year, and 52% of visits were for a diagnostic procedure. Among the 2,869 recorded rare disorders, 1,907 (66.5%) were recorded in less than 10 patients, 802 (28%) in 10 to 100 patients, 149 (5.2%) in 100 to 1,000 patients, and 11 (0.4%) in > 1,000 patients. Overall, 45.6% of individuals had no diagnosis and 6.7% had an uncertain diagnosis. Children were mainly referred by their pediatrician (46%; n = 55,755 among the 121,136 total children referrals) and adults by a medical specialist (34%; n = 14,053 among the 41,564 total adult referrals). Given the geographical coverage of the centers, the median distance from the patient's home was 25.1 km (IQR = 6.3 km-64.2 km).
Conclusions: CEMARA provides unprecedented support for epidemiological, clinical and therapeutic studies in the field of RD. Researchers can benefit from the national scope of CEMARA data, but also focus on specific diseases or patient subgroups. While this endeavor has been a major collective effort among French RD experts to gather large-scale data into a single database, it provides tremendous potential to improve patient care.
Keywords: Data warehouse; Developmental disorders; Epidemiology; Rare disease.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Ministère des Solidarités et de la Santé. https://solidarites-sante.gouv.fr/soins-et-maladies/prises-en-charge-spe.... Accessed 18 May 2021.
-
- ORPHANET. About Rare Diseases: https://www.orpha.net/consor/cgi-bin/Education_AboutRareDiseases.php?lng=EN. Accessed 18 Nov 2020.
-
- Ministère des Solidarités et de la Santé. Building Consensus and Synergies for the Registration of Rare Disease Patients in the European DGOS. About rare diseases: https://solidarites-sante.gouv.fr/soins-et-maladies/prises-en-charge-spe.... Accessed 18 Nov 2020.
-
- Ministère des Solidarités et de la Santé. The “PNMR2”. https://solidarites-sante.gouv.fr/IMG/pdf/hcsp_pnmr2_evaluation.pdf. Accessed 18 Nov 2020
-
- European Commission. The European Reference Networks by the European Commission: https://ec.europa.eu/health/ern. Accessed 18 Nov 2020.
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
