

Host adaptation in gut Firmicutes is associated with sporulation loss and altered transmission cycle
- PMID: 34348764
- PMCID: PMC8340488
- DOI: 10.1186/s13059-021-02428-6
Host adaptation in gut Firmicutes is associated with sporulation loss and altered transmission cycle
Abstract
Background: Human-to-human transmission of symbiotic, anaerobic bacteria is a fundamental evolutionary adaptation essential for membership of the human gut microbiota. However, despite its importance, the genomic and biological adaptations underpinning symbiont transmission remain poorly understood. The Firmicutes are a dominant phylum within the intestinal microbiota that are capable of producing resistant endospores that maintain viability within the environment and germinate within the intestine to facilitate transmission. However, the impact of host transmission on the evolutionary and adaptive processes within the intestinal microbiota remains unknown.
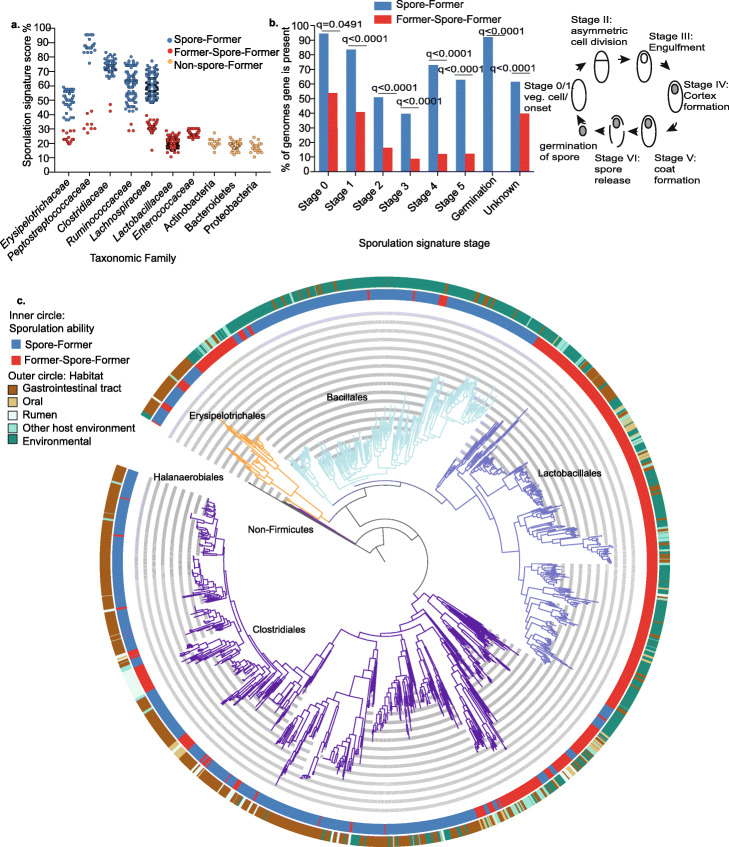
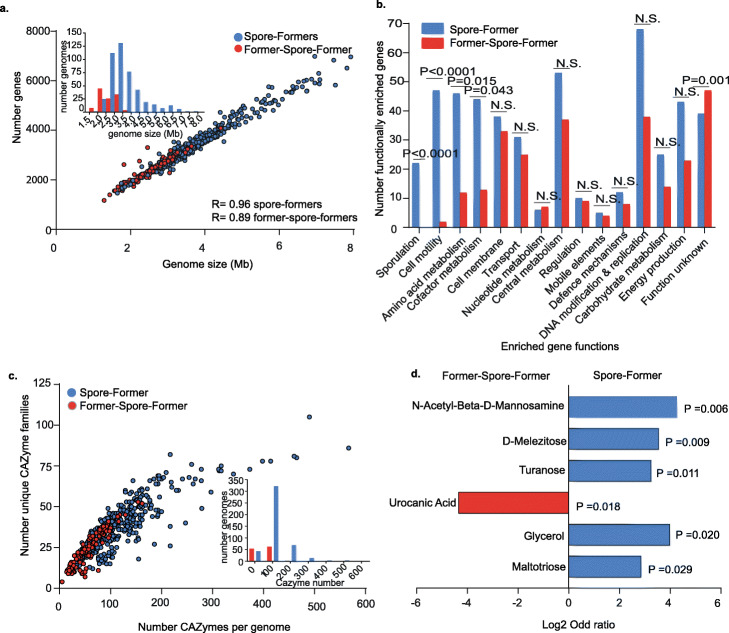
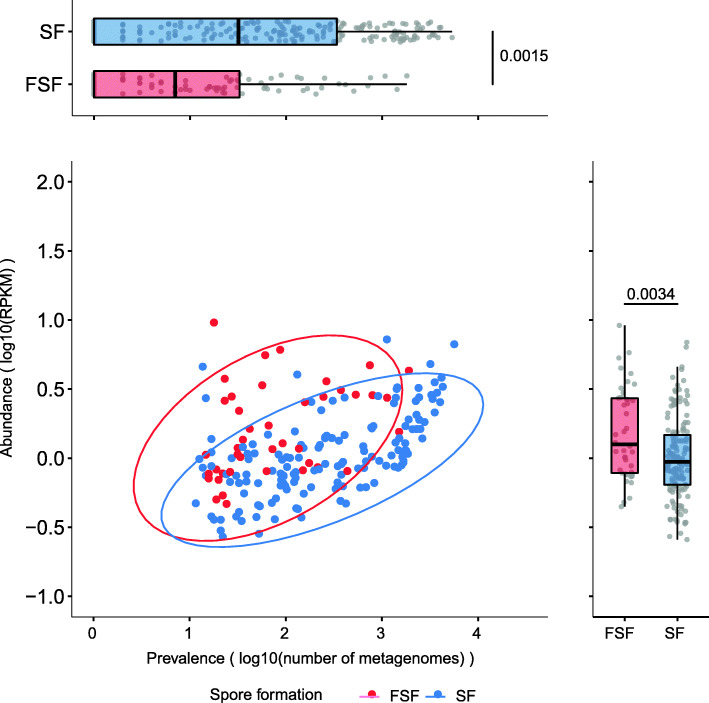
Results: We analyze 1358 genomes of Firmicutes bacteria derived from host and environment-associated habitats. Characterization of genomes as spore-forming based on the presence of sporulation-predictive genes reveals multiple losses of sporulation in many distinct lineages. Loss of sporulation in gut Firmicutes is associated with features of host-adaptation such as genome reduction and specialized metabolic capabilities. Consistent with these data, analysis of 9966 gut metagenomes from adults around the world demonstrates that bacteria now incapable of sporulation are more abundant within individuals but less prevalent in the human population compared to spore-forming bacteria.
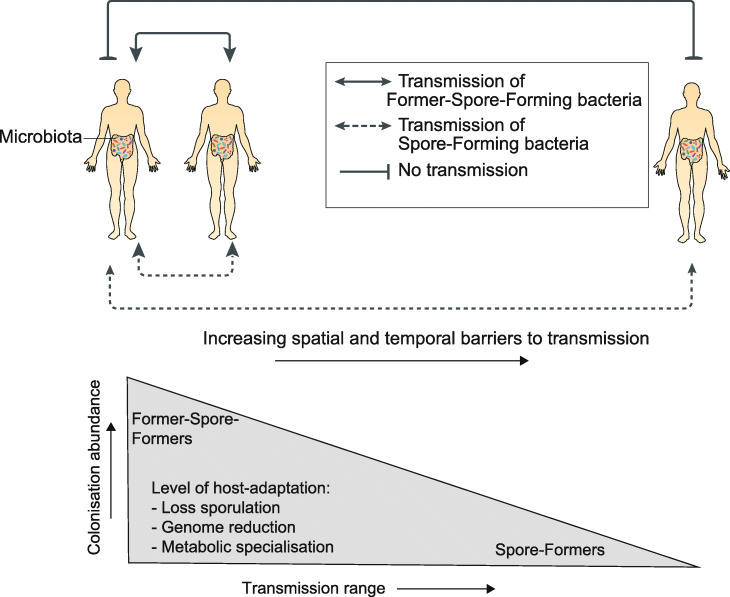
Conclusions: Our results suggest host adaptation in gut Firmicutes is an evolutionary trade-off between transmission range and colonization abundance. We reveal host transmission as an underappreciated process that shapes the evolution, assembly, and functions of gut Firmicutes.
Keywords: Bacterial transmission; Genome evolution; Genome reduction; Host adaptation; Intestinal microbiota; Metabolic specialization; Metagenomics; Microbiome; Sporulation.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
