

Phylogeny of the Varidnaviria Morphogenesis Module: Congruence and Incongruence With the Tree of Life and Viral Taxonomy
- PMID: 34349745
- PMCID: PMC8328091
- DOI: 10.3389/fmicb.2021.704052
Phylogeny of the Varidnaviria Morphogenesis Module: Congruence and Incongruence With the Tree of Life and Viral Taxonomy
Abstract
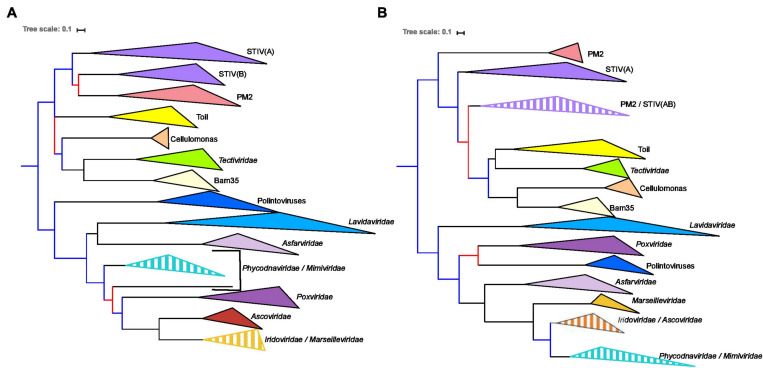
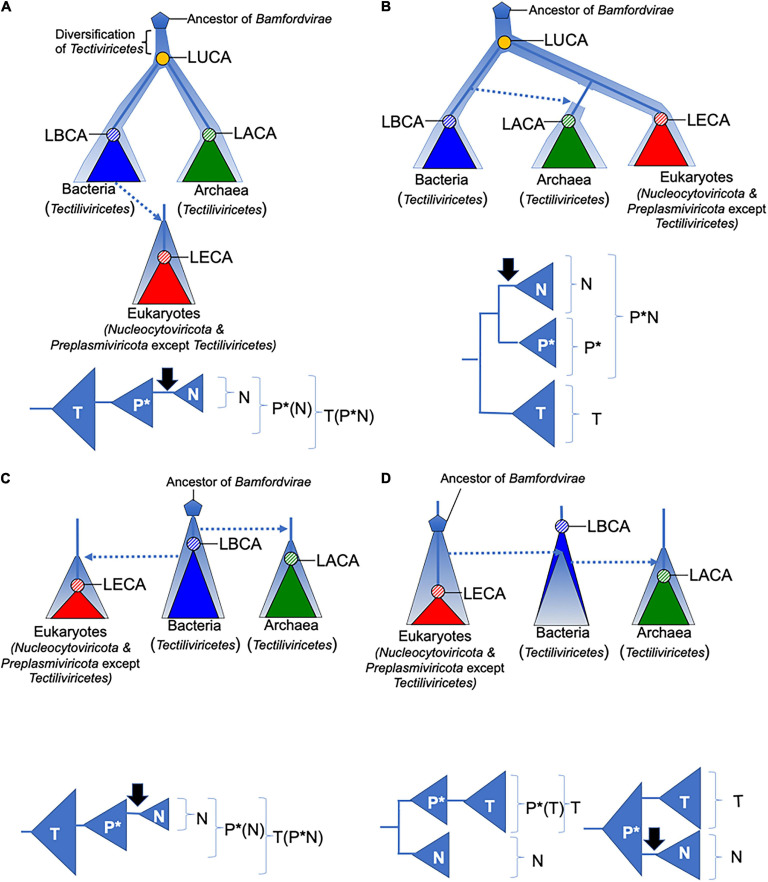
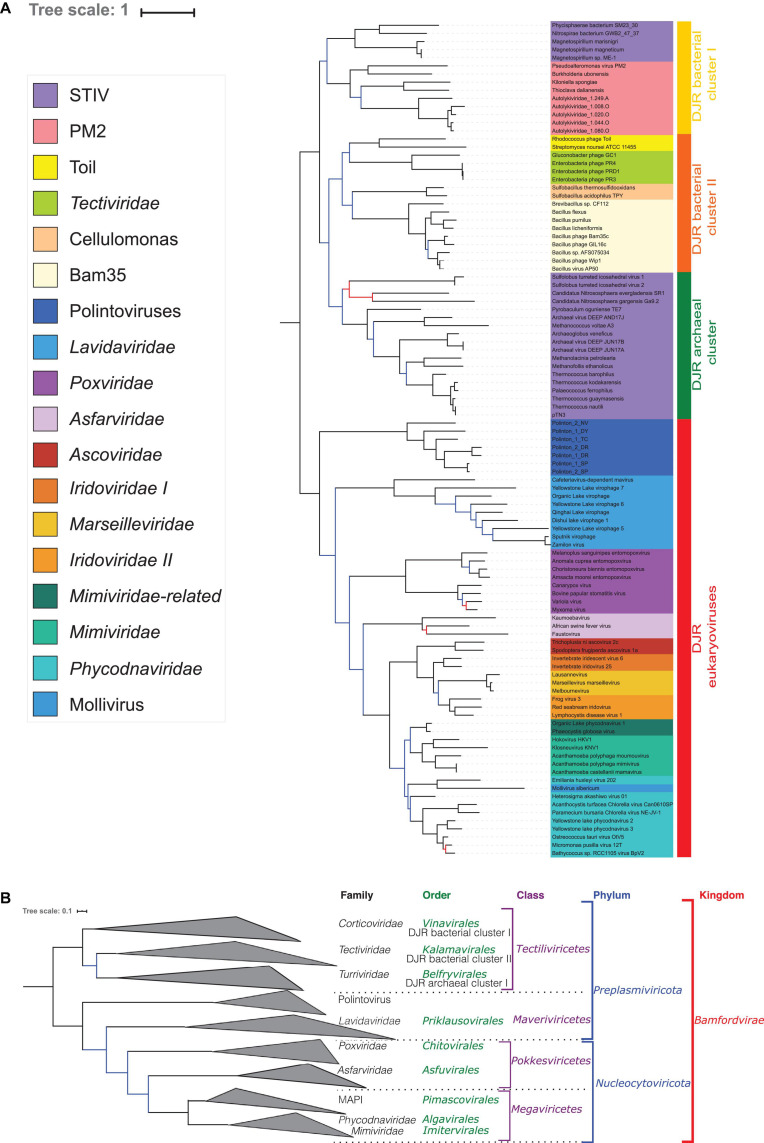
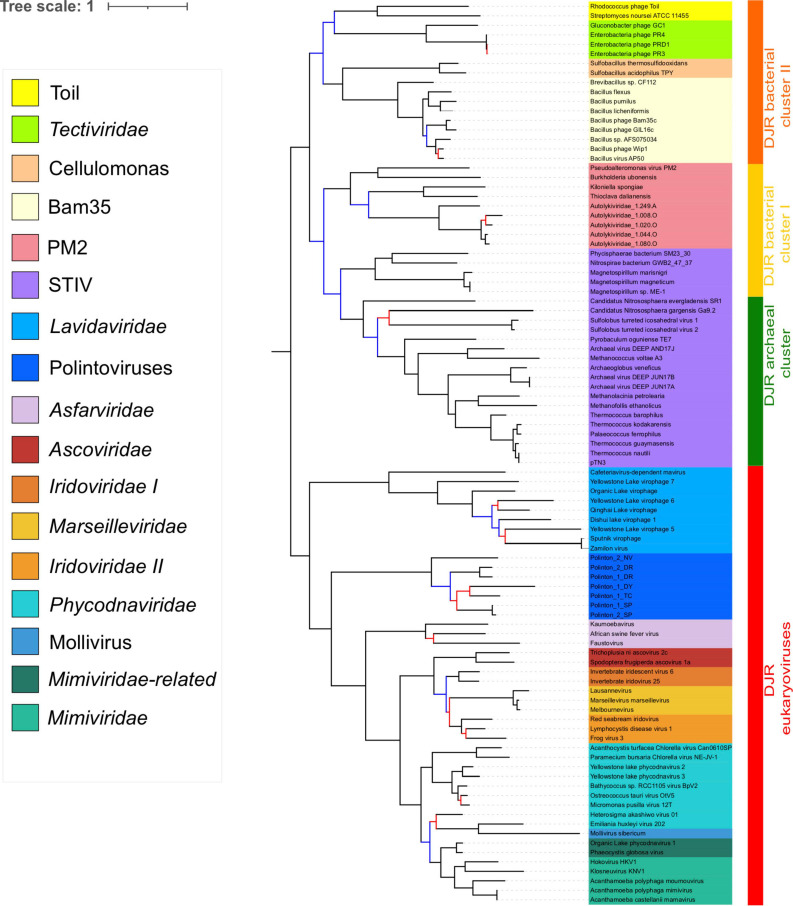
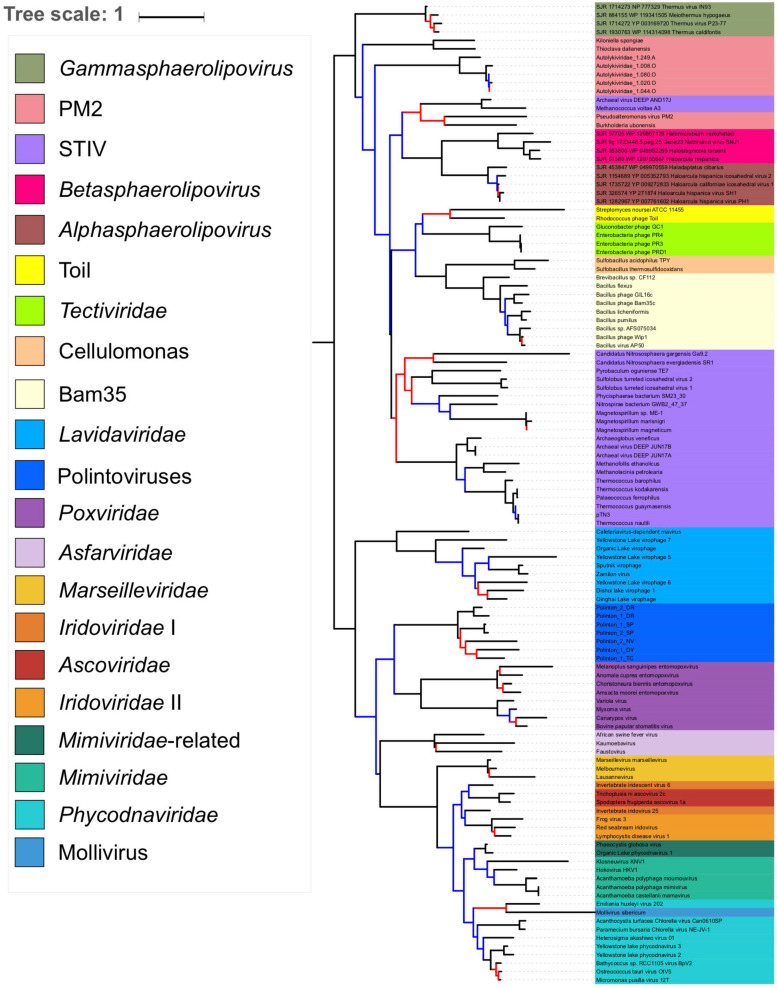
Double-stranded DNA viruses of the realm Varidnaviria (formerly PRD1-adenovirus lineage) are characterized by homologous major capsid proteins (MCPs) containing one (kingdom: Helvetiavirae) or two β-barrel domains (kingdom: Bamfordvirae) known as the jelly roll folds. Most of them also share homologous packaging ATPases (pATPases). Remarkably, Varidnaviria infect hosts from the three domains of life, suggesting that these viruses could be very ancient and share a common ancestor. Here, we analyzed the evolutionary history of Varidnaviria based on single and concatenated phylogenies of their MCPs and pATPases. We excluded Adenoviridae from our analysis as their MCPs and pATPases are too divergent. Sphaerolipoviridae, the only family in the kingdom Helvetiavirae, exhibit a complex history: their MCPs are very divergent from those of other Varidnaviria, as expected, but their pATPases groups them with Bamfordvirae. In single and concatenated trees, Bamfordvirae infecting archaea were grouped with those infecting bacteria, in contradiction with the cellular tree of life, whereas those infecting eukaryotes were organized into three monophyletic groups: the Nucleocytoviricota phylum, formerly known as the Nucleo-Cytoplasmic Large DNA Viruses (NCLDVs), Lavidaviridae (virophages) and Polintoviruses. Although our analysis mostly supports the recent classification proposed by the International Committee on Taxonomy of Viruses (ICTV), it also raises questions, such as the validity of the Adenoviridae and Helvetiavirae ranking. Based on our phylogeny, we discuss current hypotheses on the origin and evolution of Varidnaviria and suggest new ones to reconcile the viral and cellular trees.
Keywords: NCLDV; dsDNA viruses; evolution; giant viruses; viral taxonomy.
Copyright © 2021 Woo, Gaia, Guglielmini, Da Cunha and Forterre.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Cellular homologs of the double jelly-roll major capsid proteins clarify the origins of an ancient virus kingdom.Proc Natl Acad Sci U S A. 2022 Feb 1;119(5):e2120620119. doi: 10.1073/pnas.2120620119. Proc Natl Acad Sci U S A. 2022. PMID: 35078938 Free PMC article.
-
Natural history of eukaryotic DNA viruses with double jelly-roll major capsid proteins.Proc Natl Acad Sci U S A. 2024 Jun 4;121(23):e2405771121. doi: 10.1073/pnas.2405771121. Epub 2024 May 28. Proc Natl Acad Sci U S A. 2024. PMID: 38805295 Free PMC article.
-
Natural history of eukaryotic DNA viruses with double jelly-roll major capsid proteins.bioRxiv [Preprint]. 2024 Mar 18:2024.03.18.585575. doi: 10.1101/2024.03.18.585575. bioRxiv. 2024. Update in: Proc Natl Acad Sci U S A. 2024 Jun 4;121(23):e2405771121. doi: 10.1073/pnas.2405771121. PMID: 38712159 Free PMC article. Updated. Preprint.
-
Evolutionary genomics of nucleo-cytoplasmic large DNA viruses.Virus Res. 2006 Apr;117(1):156-84. doi: 10.1016/j.virusres.2006.01.009. Epub 2006 Feb 21. Virus Res. 2006. PMID: 16494962 Review.
-
HCIV-1 and Other Tailless Icosahedral Internal Membrane-Containing Viruses of the Family Sphaerolipoviridae.Viruses. 2017 Feb 18;9(2):32. doi: 10.3390/v9020032. Viruses. 2017. PMID: 28218714 Free PMC article. Review.
Cited by
-
A billion years arms-race between viruses, virophages, and eukaryotes.Elife. 2023 Jun 26;12:RP86617. doi: 10.7554/eLife.86617. Elife. 2023. PMID: 37358563 Free PMC article.
-
Jorvik: A membrane-containing phage that will likely found a new family within Vinavirales.iScience. 2023 Sep 29;26(11):108104. doi: 10.1016/j.isci.2023.108104. eCollection 2023 Nov 17. iScience. 2023. PMID: 37867962 Free PMC article.
-
Genomic insights reveal community structure and phylogenetic associations of endohyphal bacteria and viruses in fungal endophytes.Environ Microbiome. 2025 Jul 25;20(1):95. doi: 10.1186/s40793-025-00757-8. Environ Microbiome. 2025. PMID: 40713930 Free PMC article.
-
A critical analysis of the current state of virus taxonomy.Front Microbiol. 2023 Aug 3;14:1240993. doi: 10.3389/fmicb.2023.1240993. eCollection 2023. Front Microbiol. 2023. PMID: 37601376 Free PMC article. Review.
-
Viral Complexity.Biomolecules. 2022 Jul 30;12(8):1061. doi: 10.3390/biom12081061. Biomolecules. 2022. PMID: 36008955 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources