

SCITO-seq: single-cell combinatorial indexed cytometry sequencing
- PMID: 34354295
- PMCID: PMC8643207
- DOI: 10.1038/s41592-021-01222-3
SCITO-seq: single-cell combinatorial indexed cytometry sequencing
Abstract
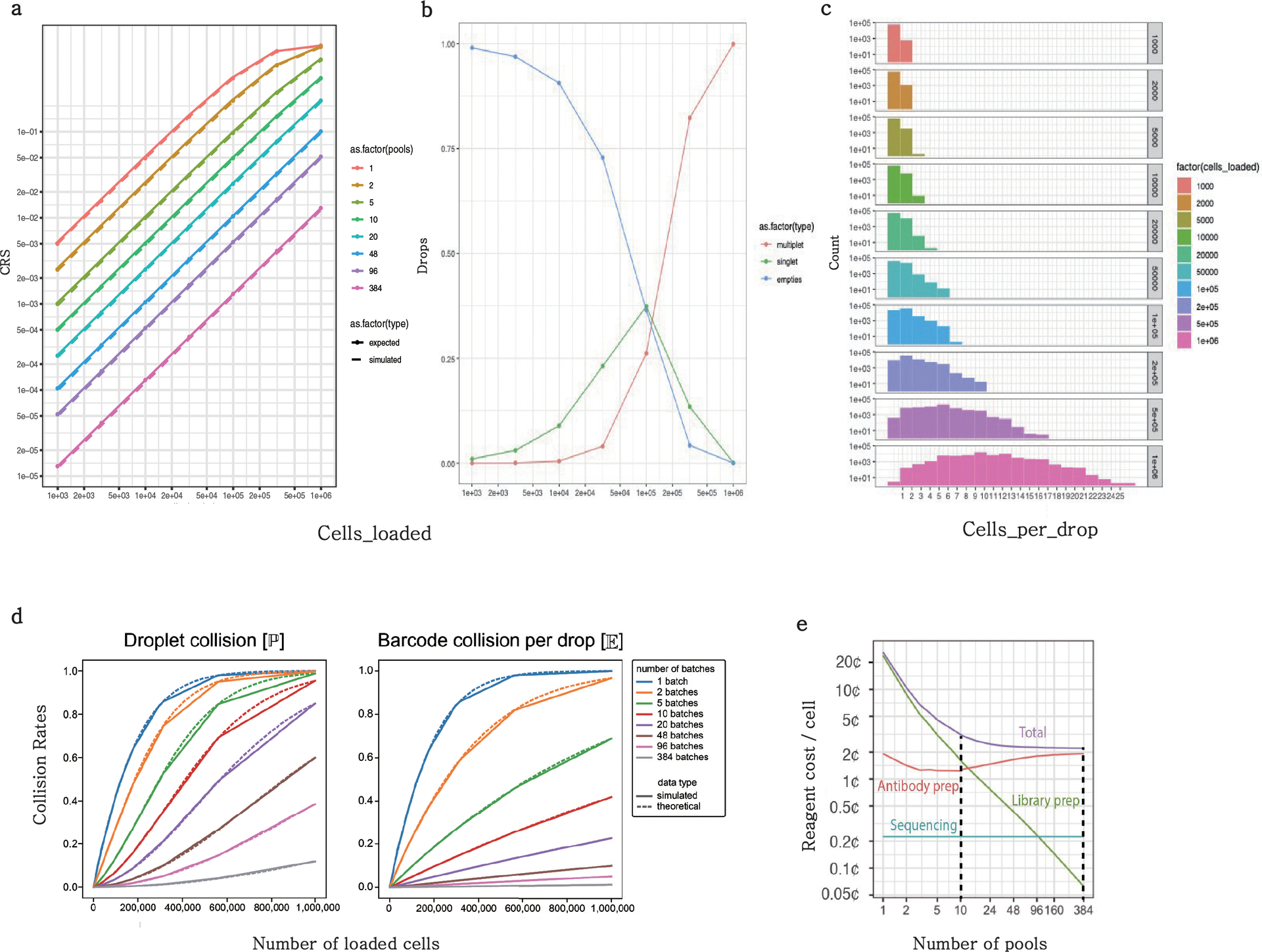
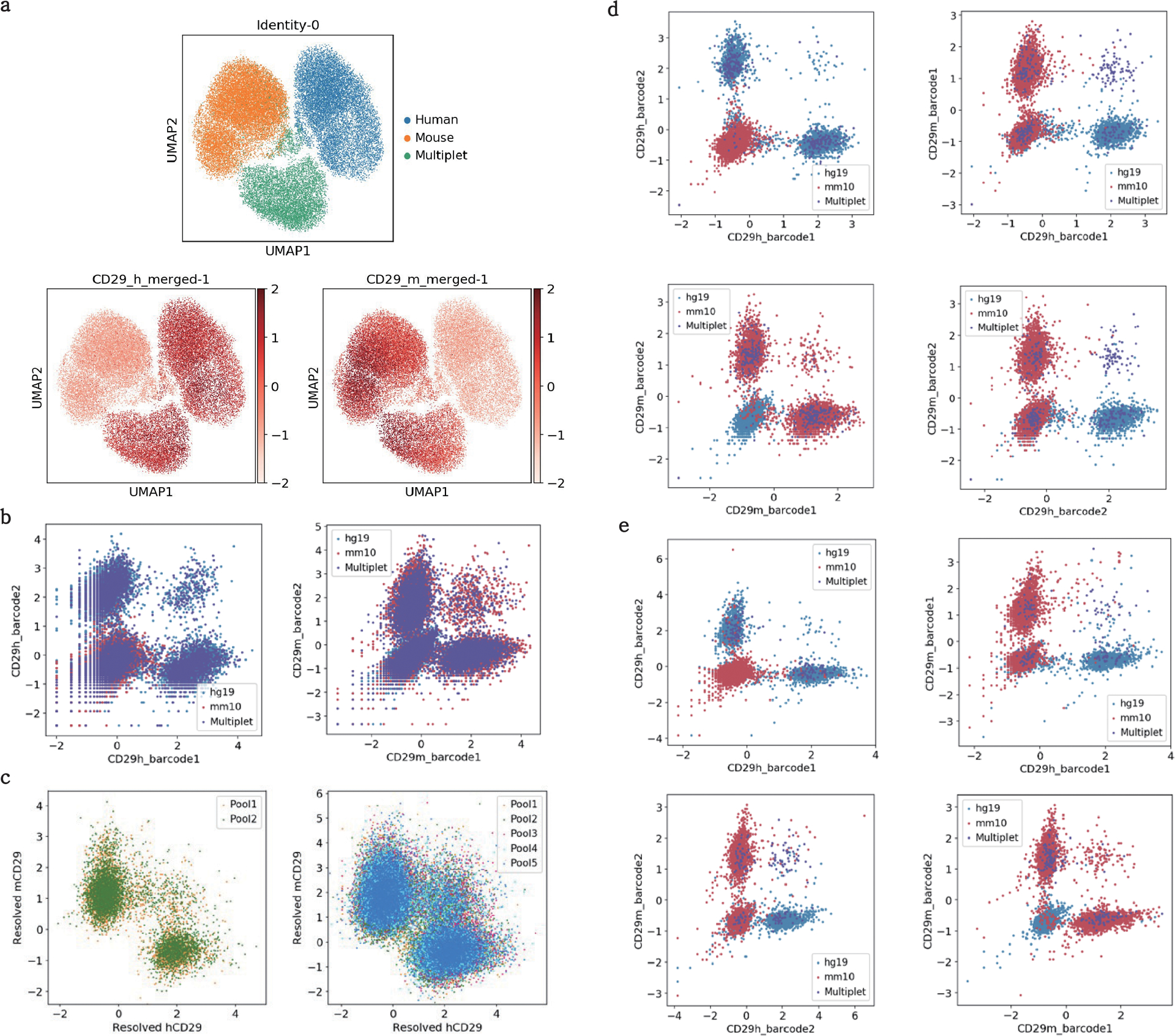
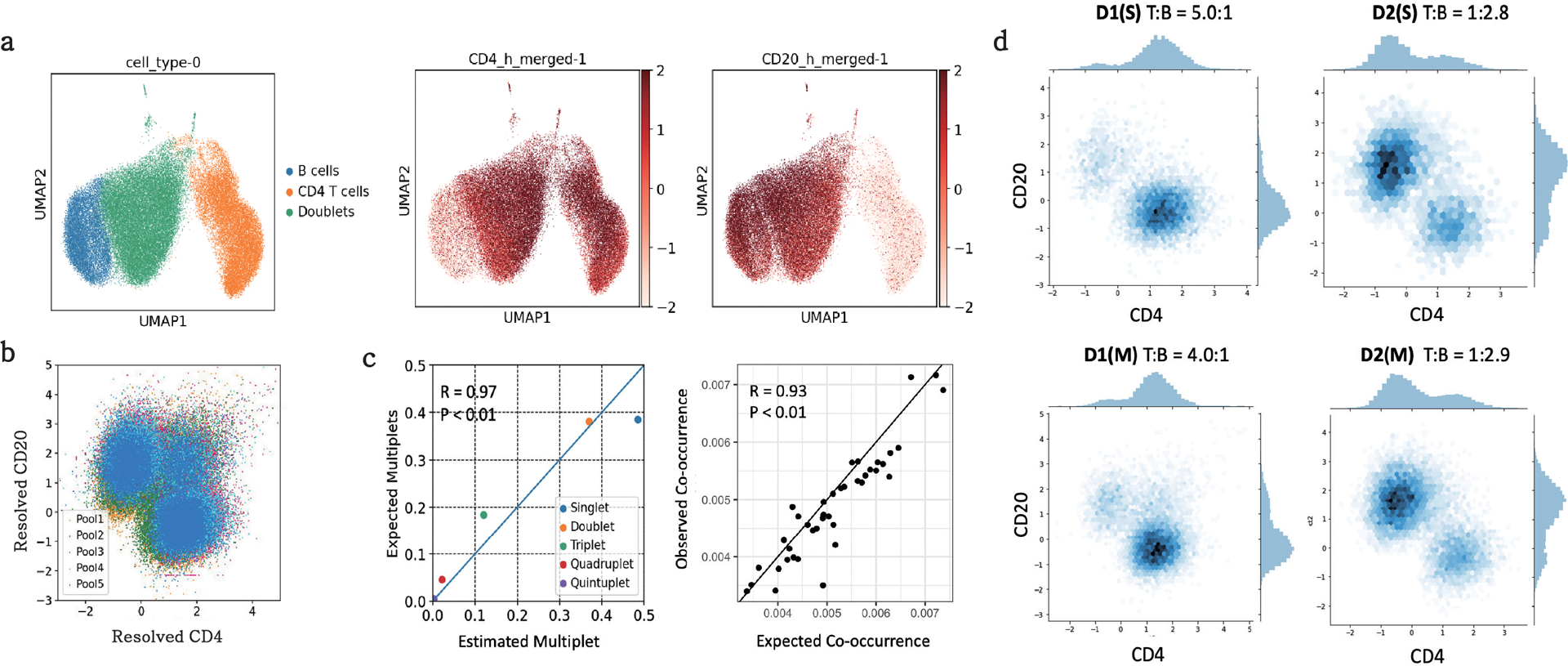
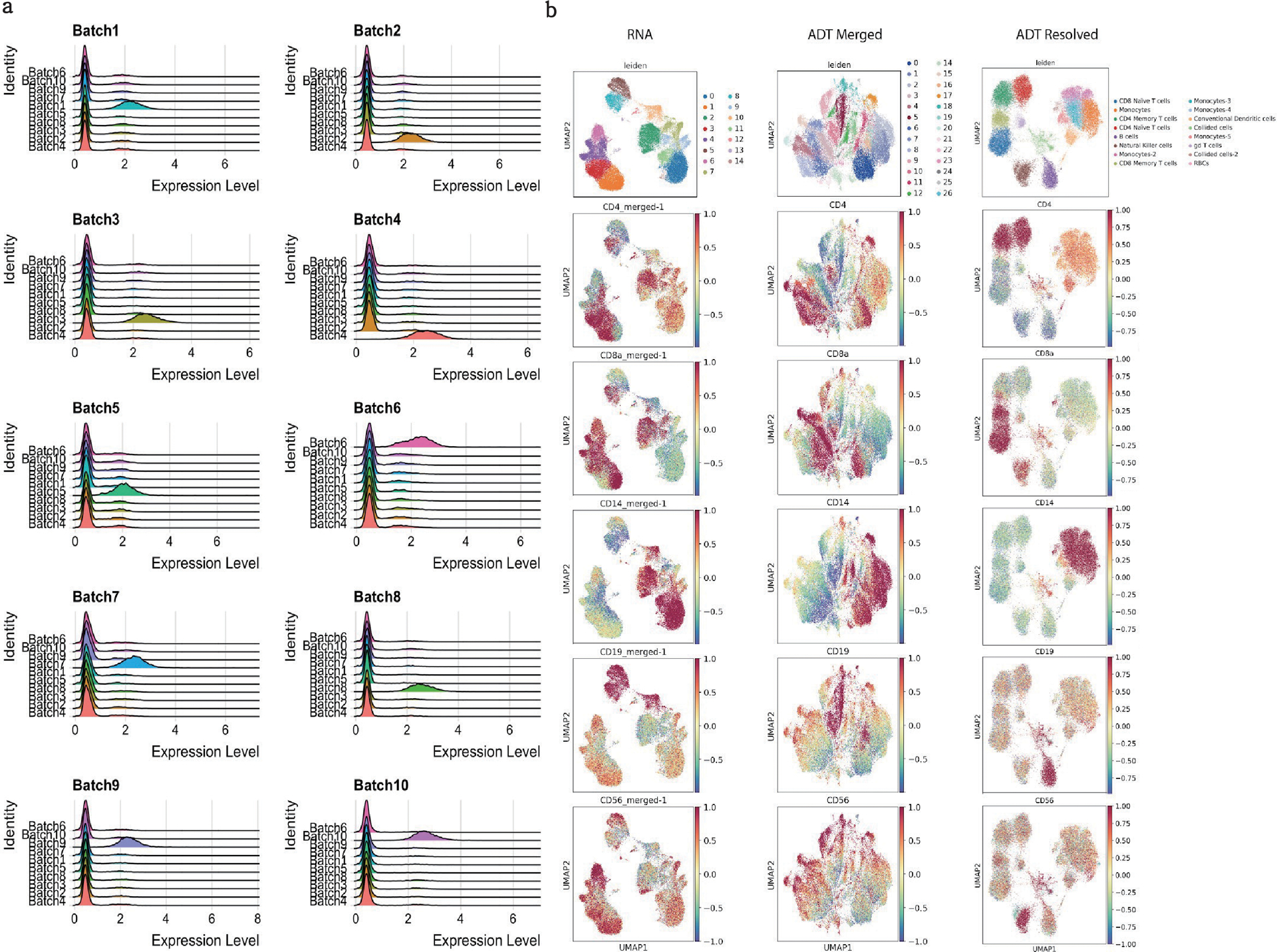
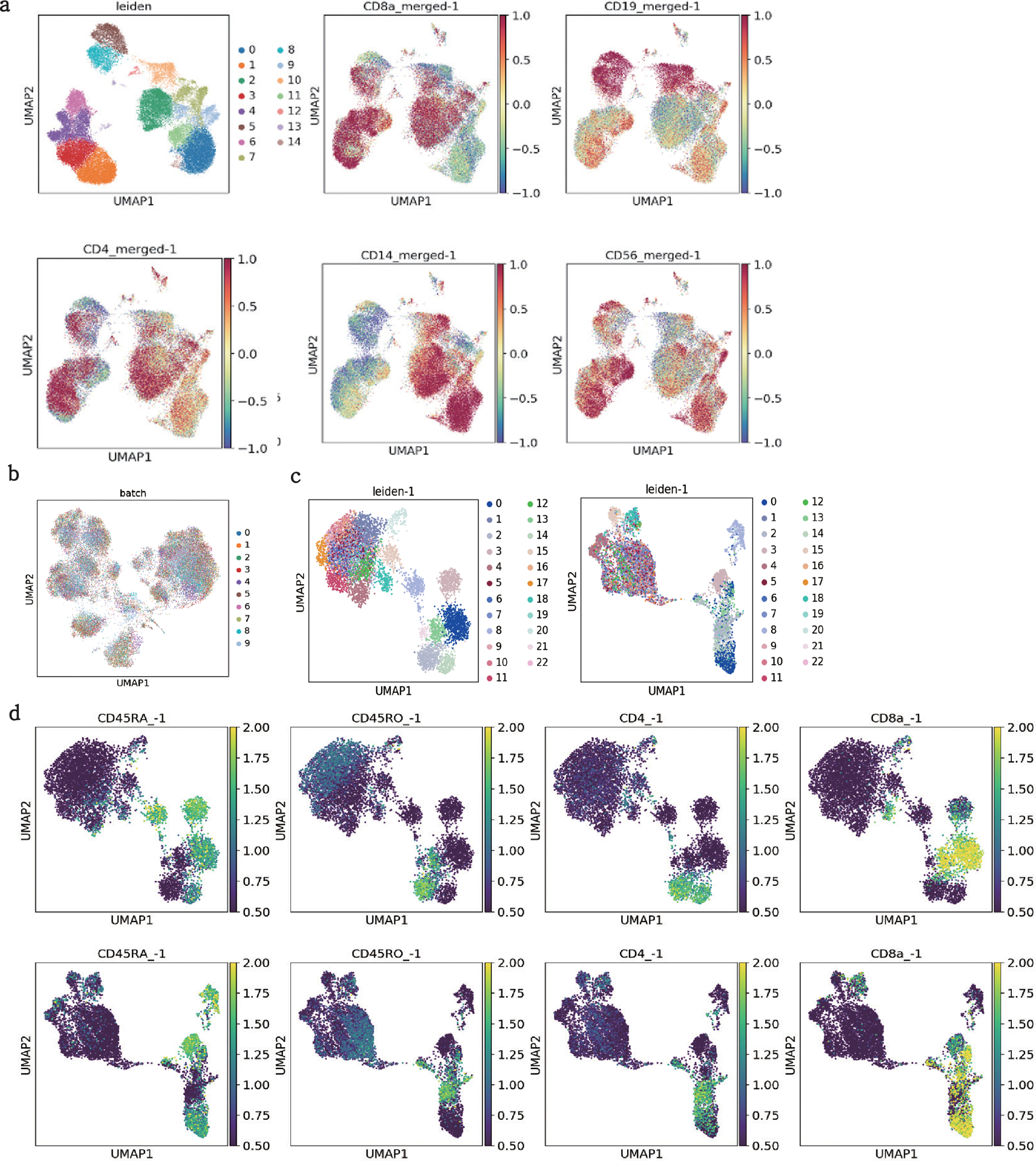
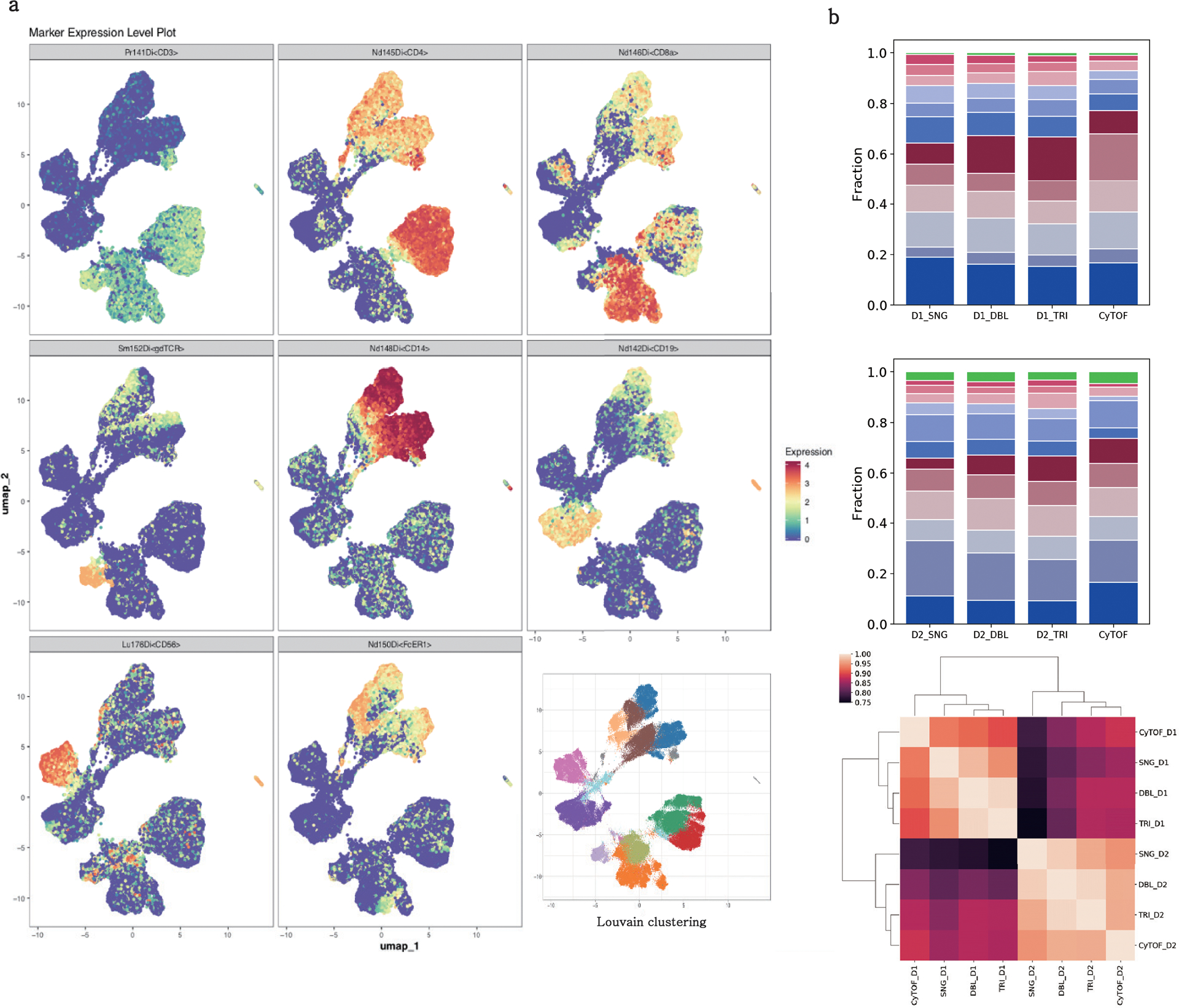
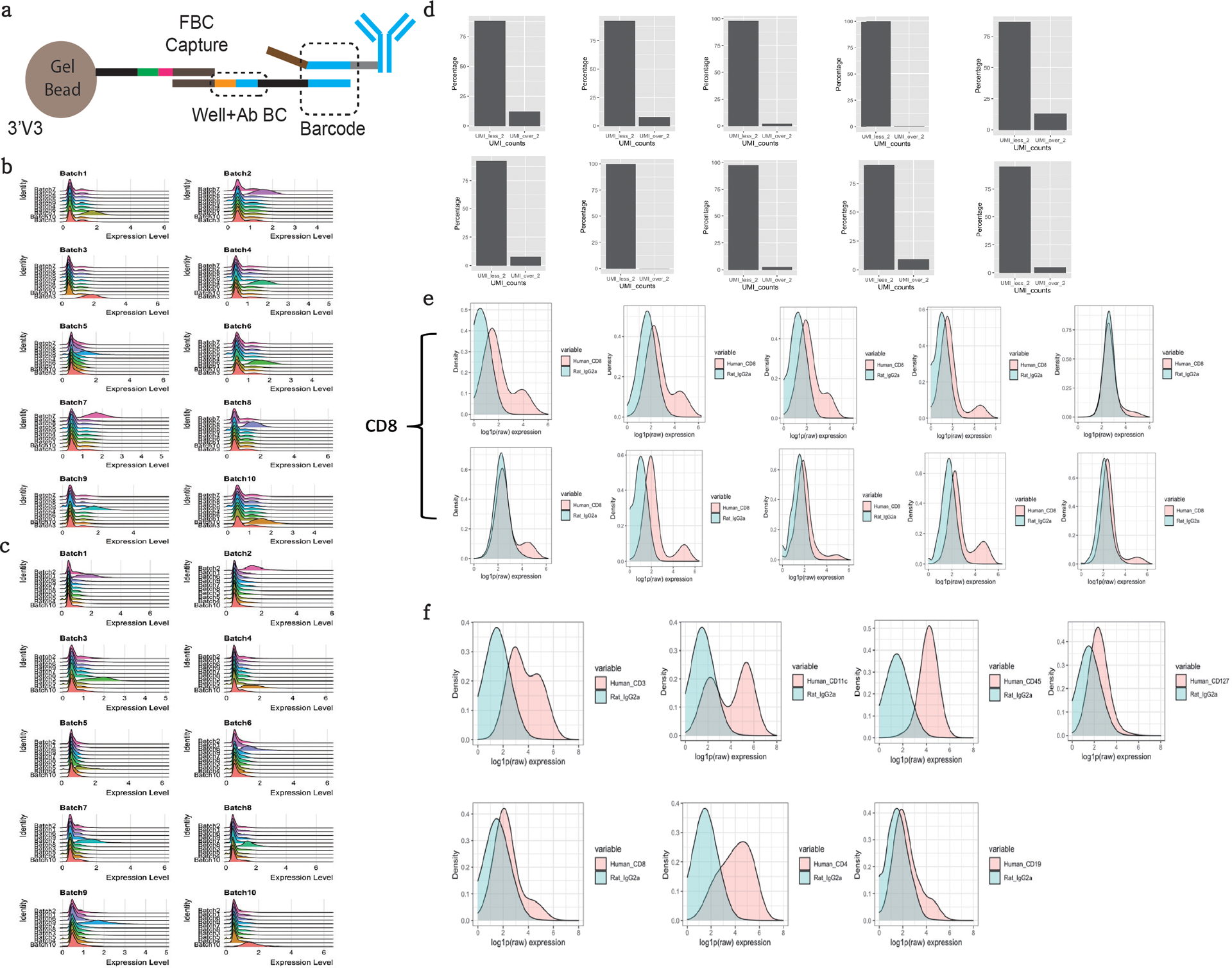
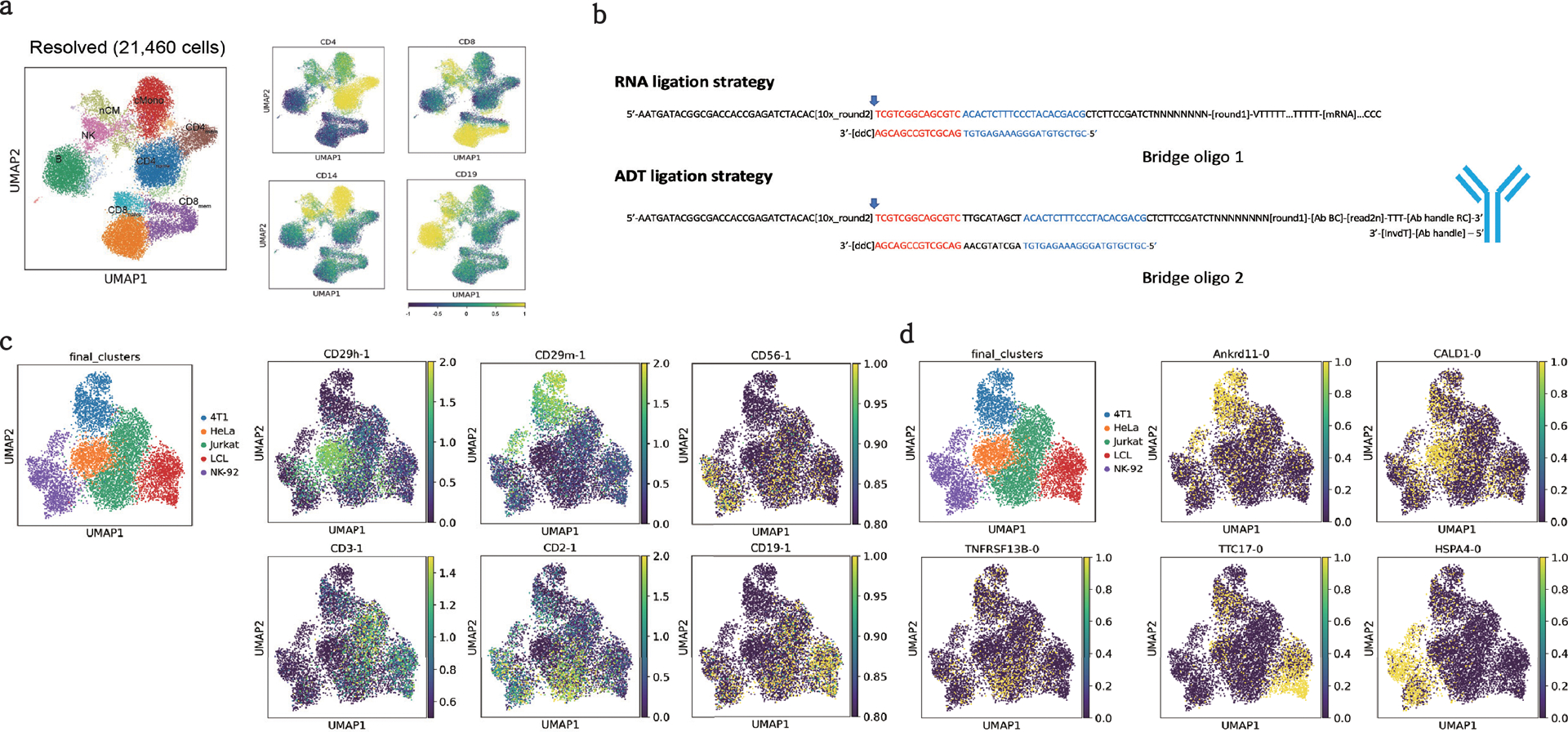
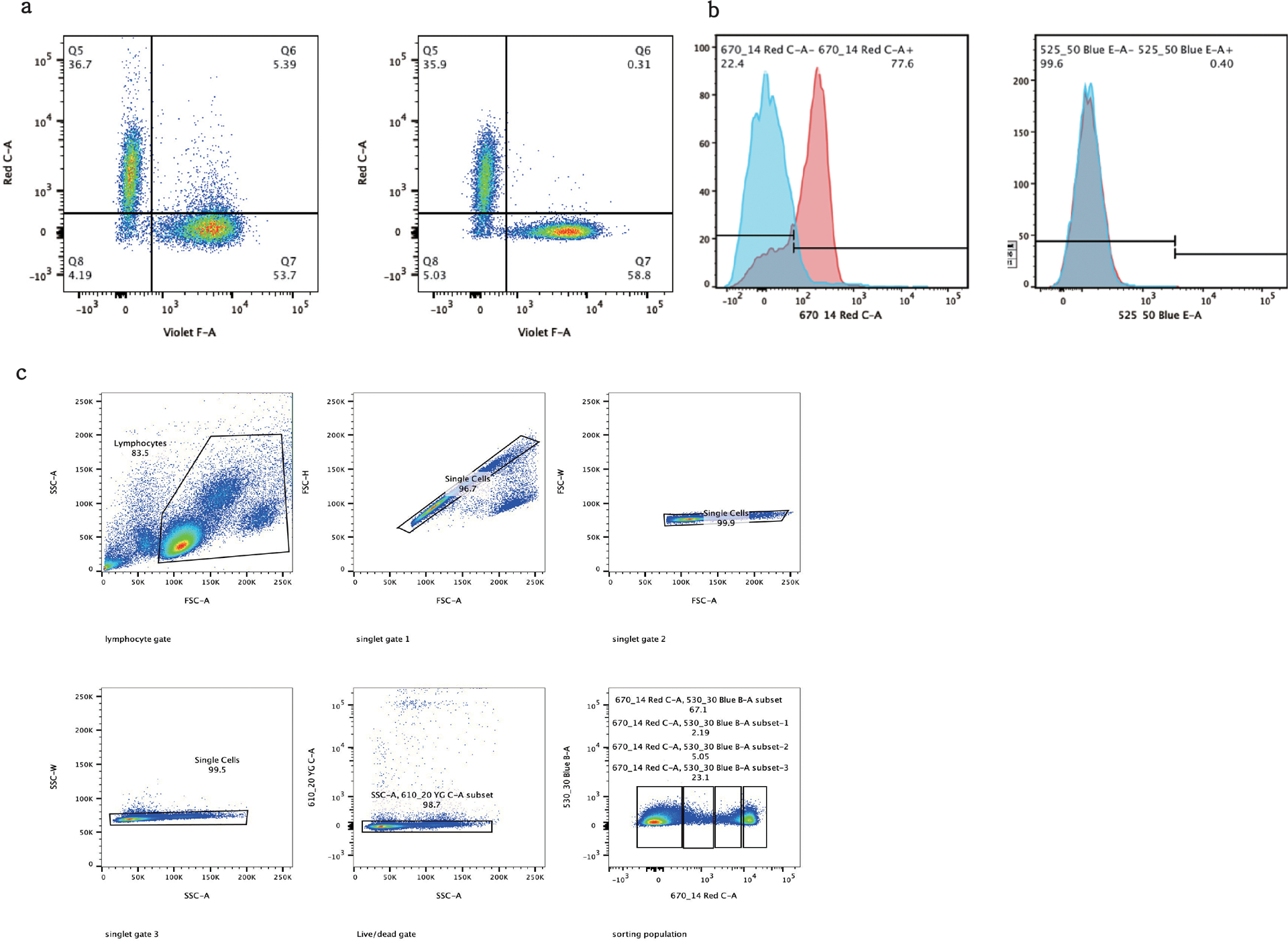
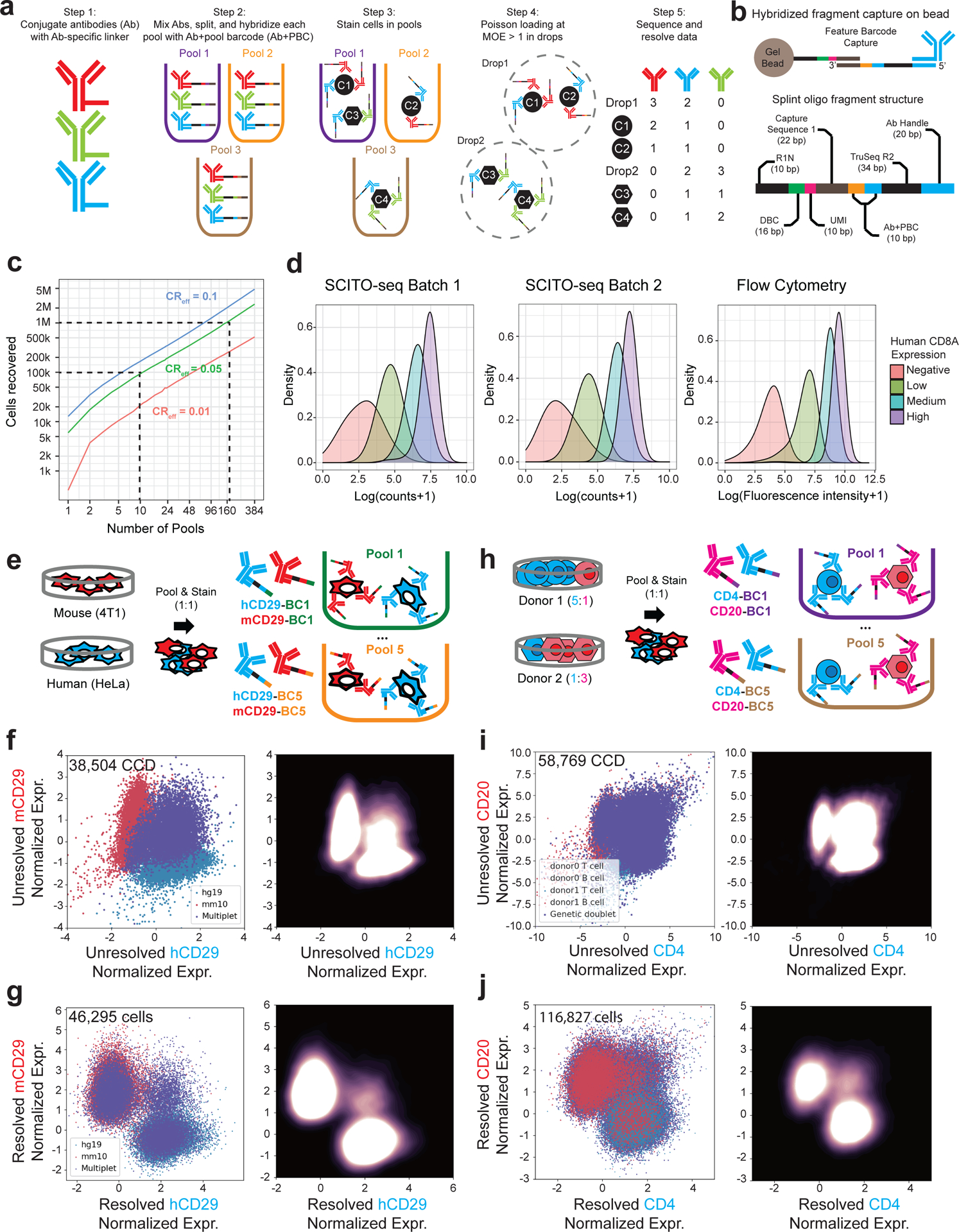
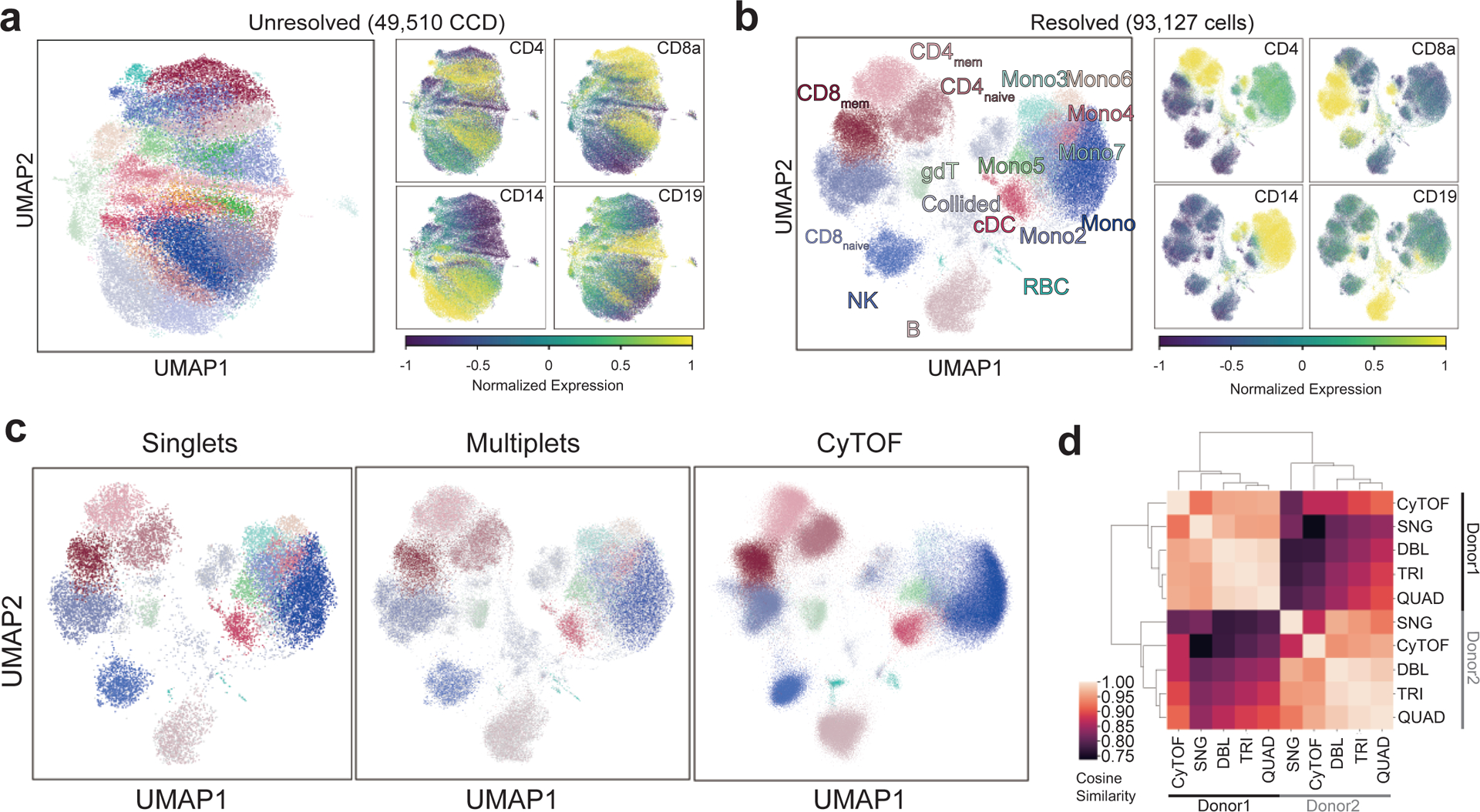
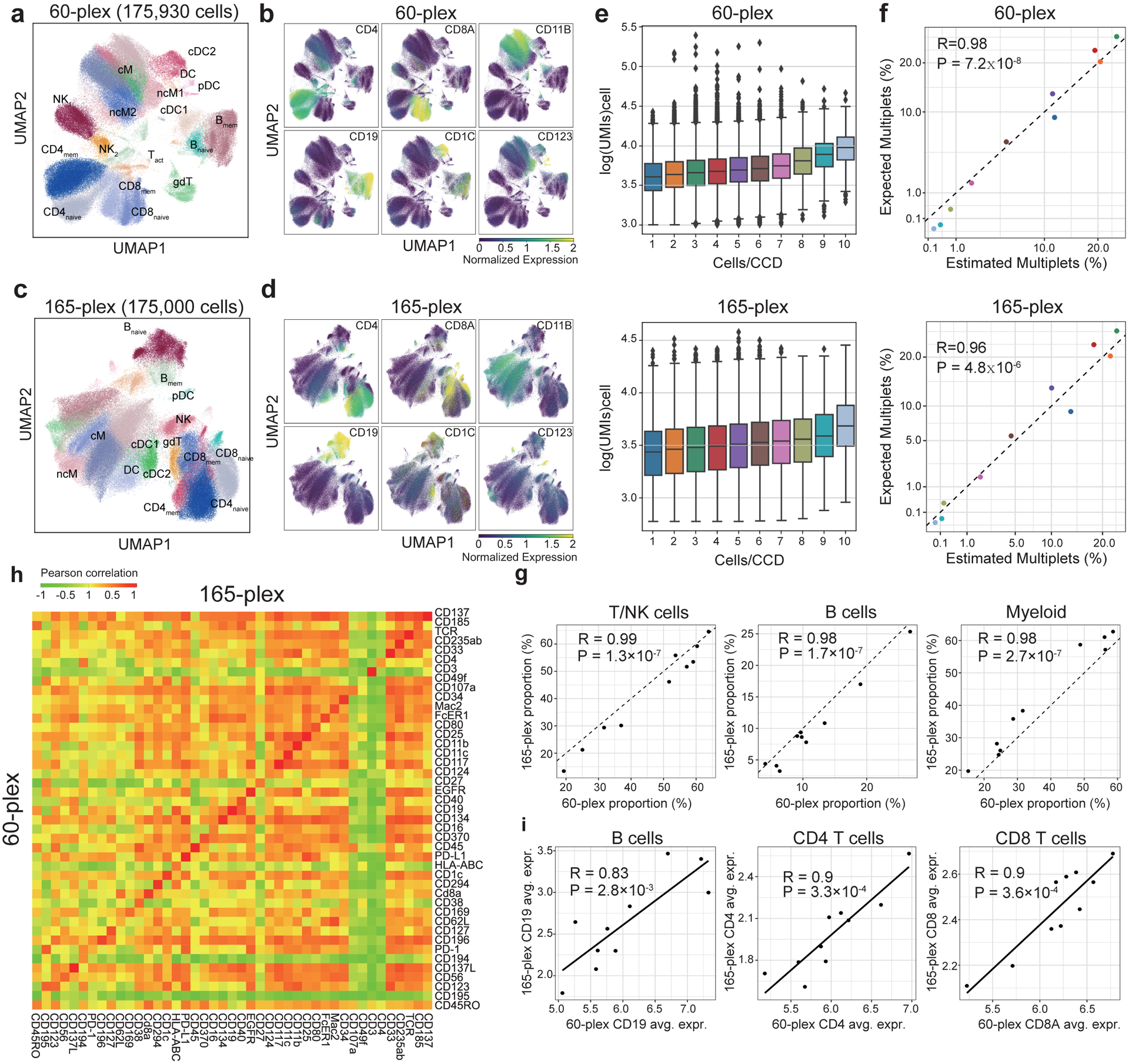
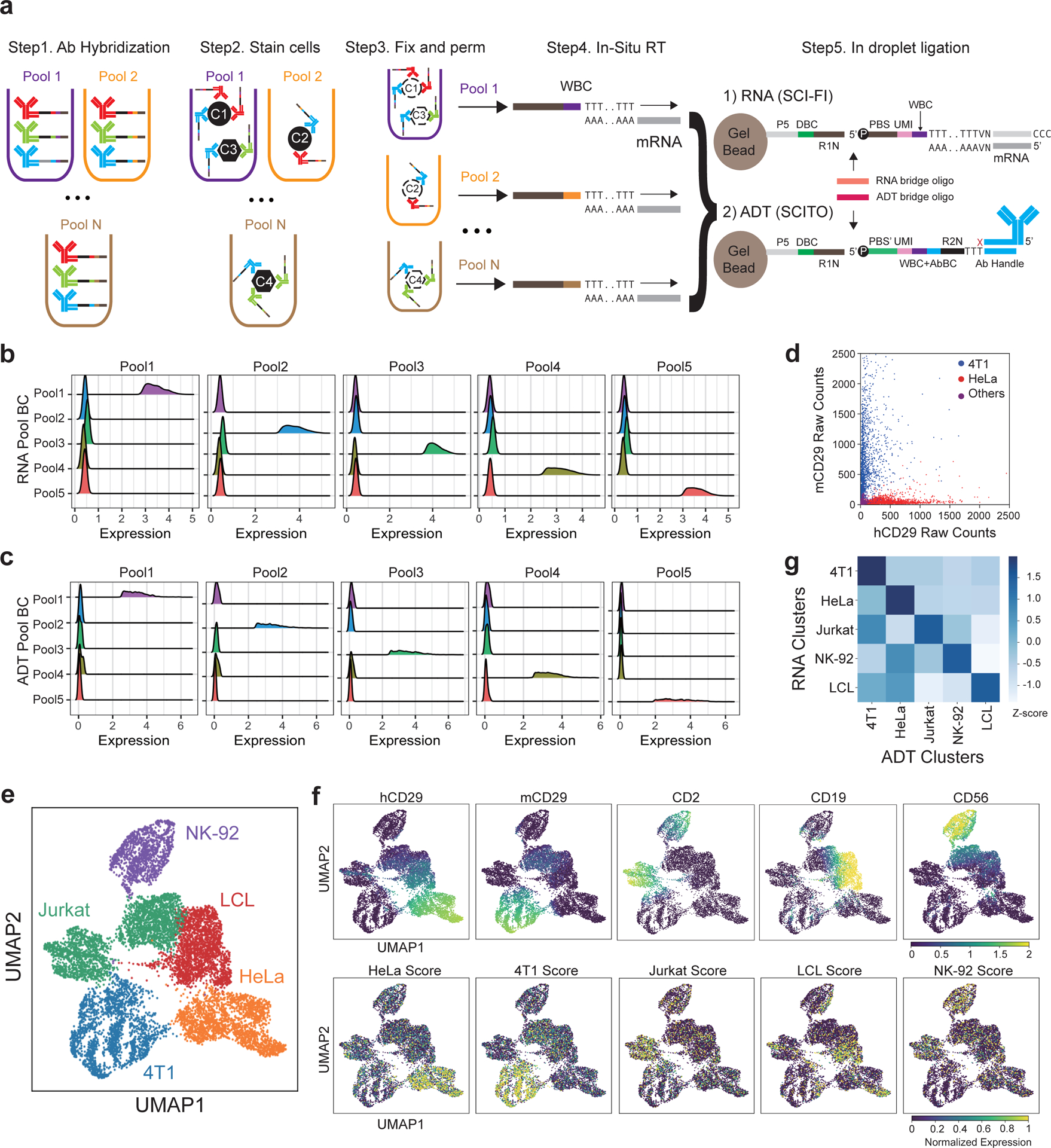
The development of DNA-barcoded antibodies to tag cell surface molecules has enabled the use of droplet-based single-cell sequencing (dsc-seq) to profile protein abundances from thousands of cells simultaneously. As compared to flow and mass cytometry, the high per cell cost of current dsc-seq-based workflows precludes their use in clinical applications and large-scale pooled screens. Here, we introduce SCITO-seq, a workflow that uses splint oligonucleotides (oligos) to enable combinatorially indexed dsc-seq of DNA-barcoded antibodies from over 105 cells per reaction using commercial microfluidics. By encoding sample barcodes into splint oligos, we demonstrate that multiplexed SCITO-seq produces reproducible estimates of cellular composition and surface protein expression comparable to those from mass cytometry. We further demonstrate two modified splint oligo designs that extend SCITO-seq to achieve compatibility with commercial DNA-barcoded antibodies and simultaneous expression profiling of the transcriptome and surface proteins from the same cell. These results demonstrate SCITO-seq as a flexible and ultra-high-throughput platform for sequencing-based single-cell protein and multimodal profiling.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures

References
MAIN REFERENCES
METHOD REFERENCES
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
