

Synthesis and pharmacological evaluation of [18F]PBR316: a novel PET ligand targeting the translocator protein 18 kDa (TSPO) with low binding sensitivity to human single nucleotide polymorphism rs6971
- PMID: 34355185
- PMCID: PMC8292990
- DOI: 10.1039/d1md00035g
Synthesis and pharmacological evaluation of [18F]PBR316: a novel PET ligand targeting the translocator protein 18 kDa (TSPO) with low binding sensitivity to human single nucleotide polymorphism rs6971
Abstract
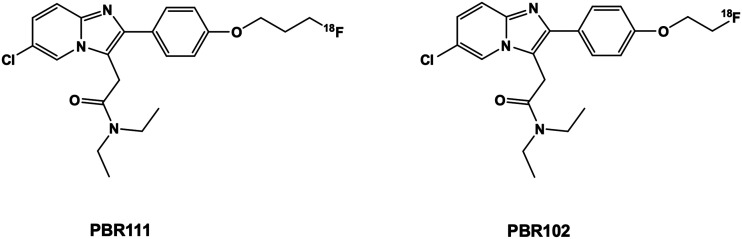
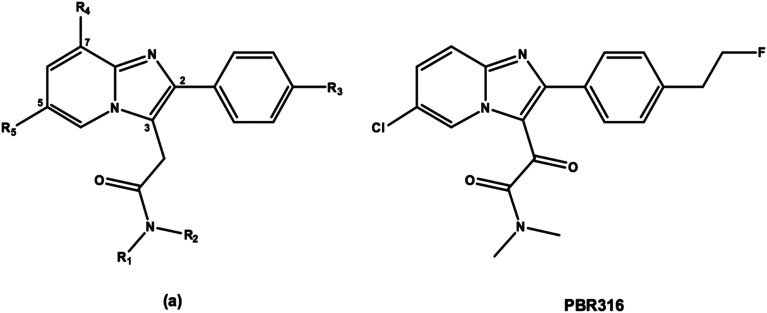
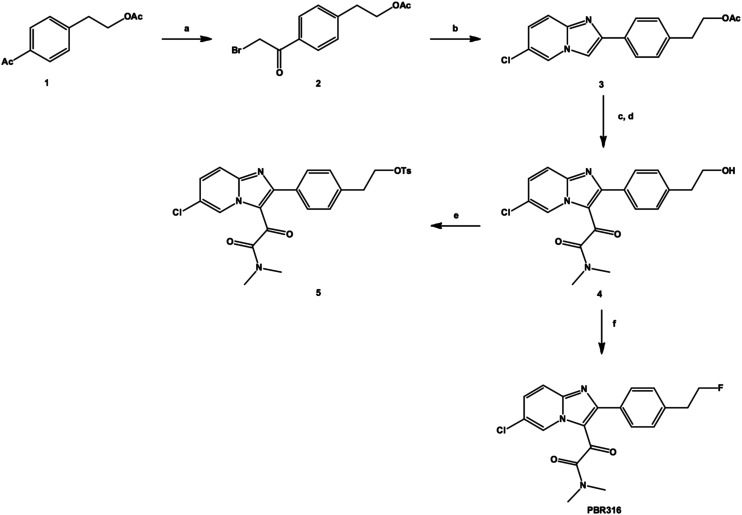
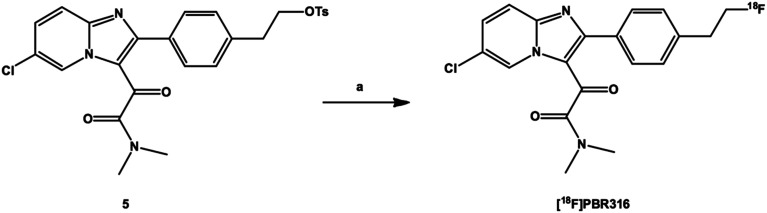
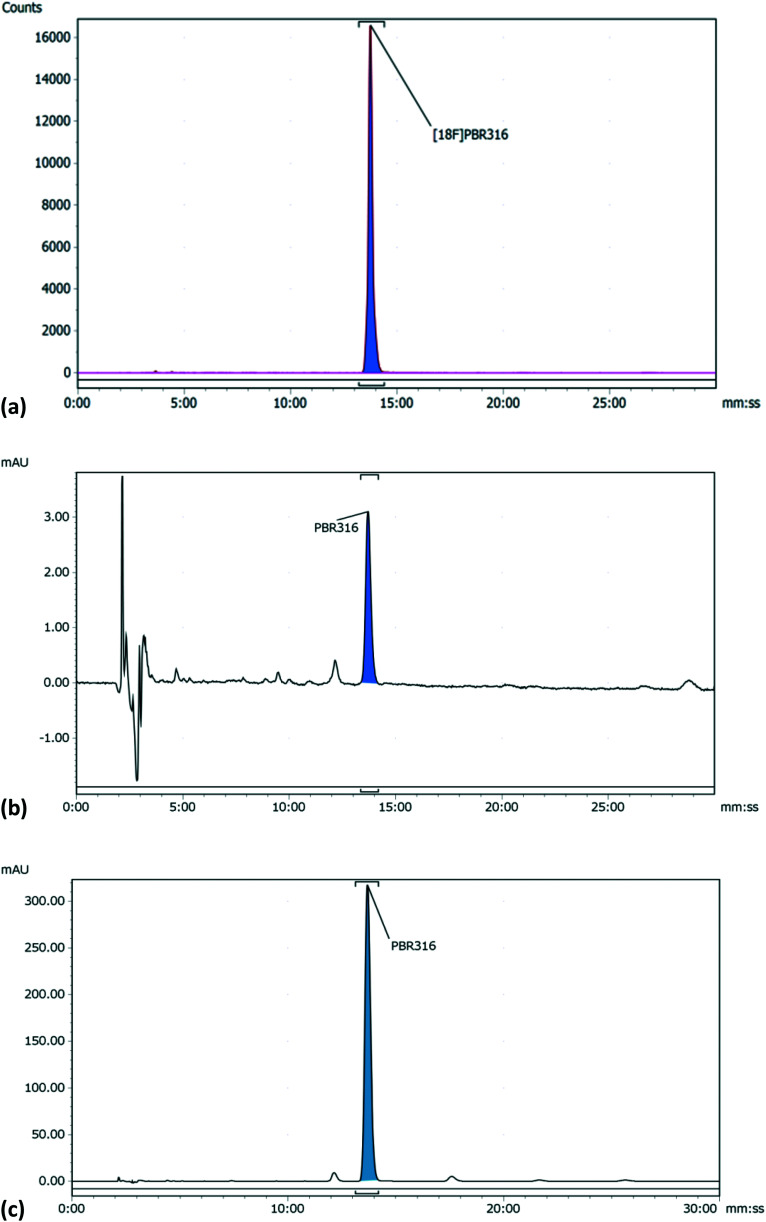
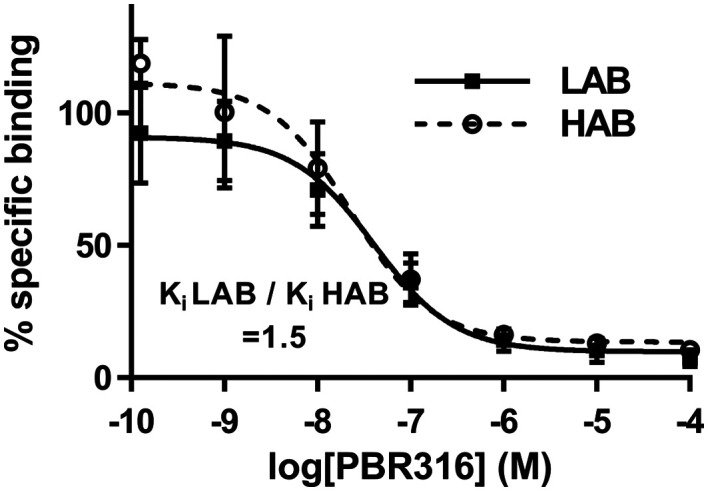
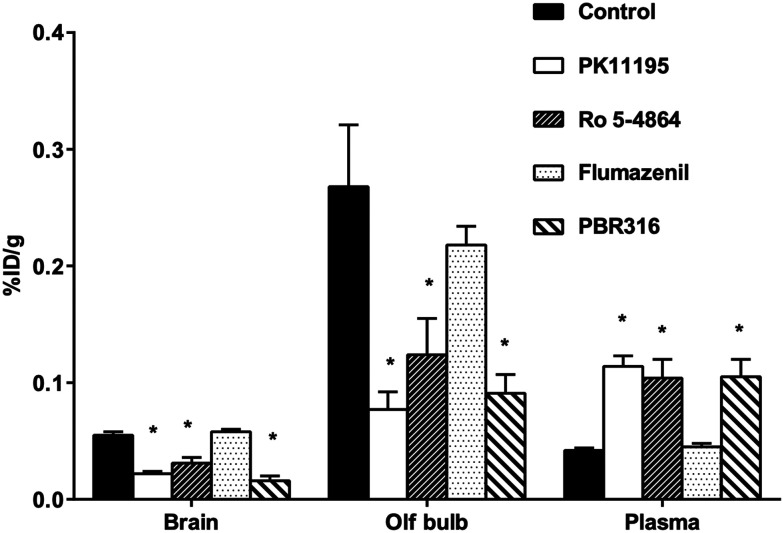
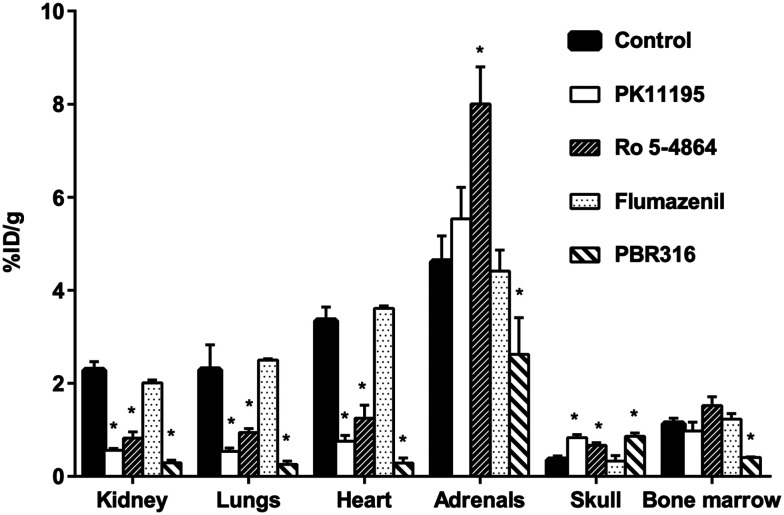
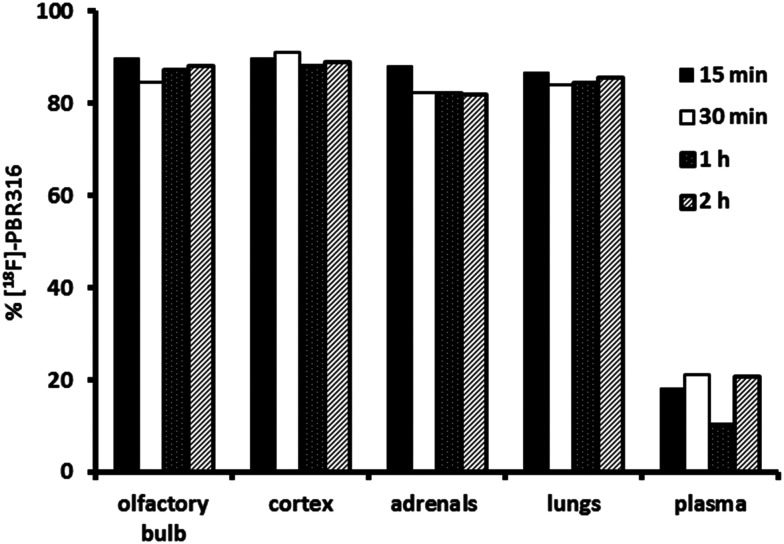
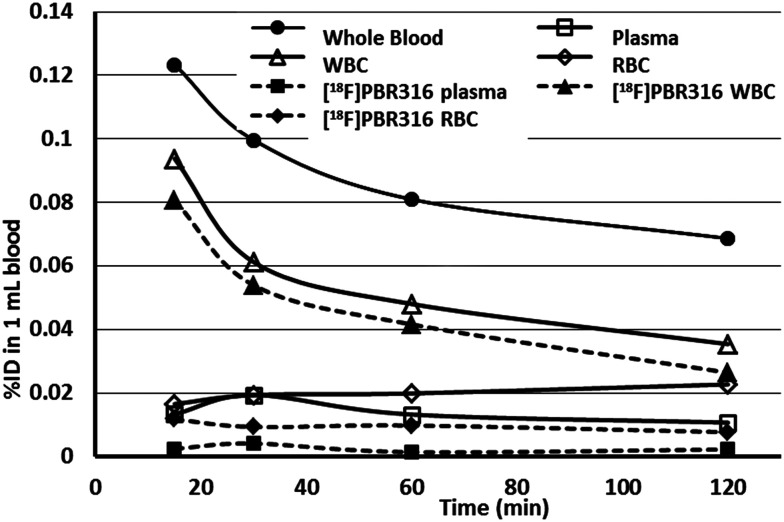
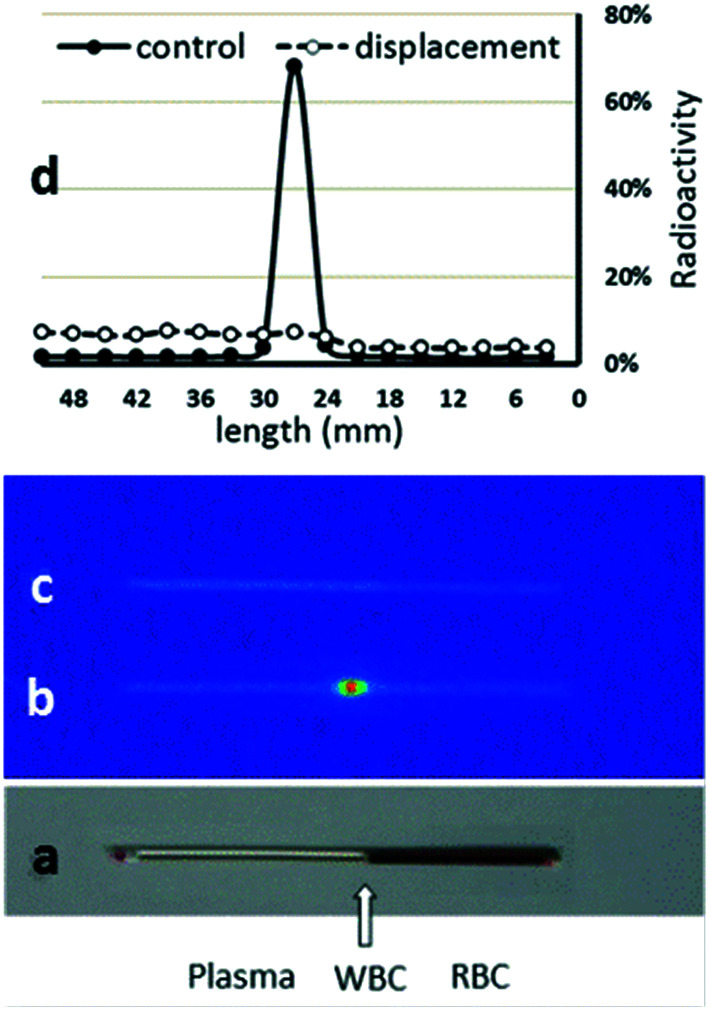
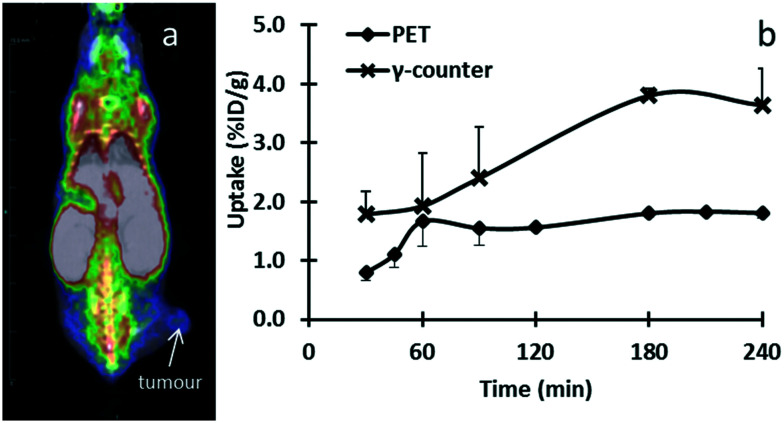
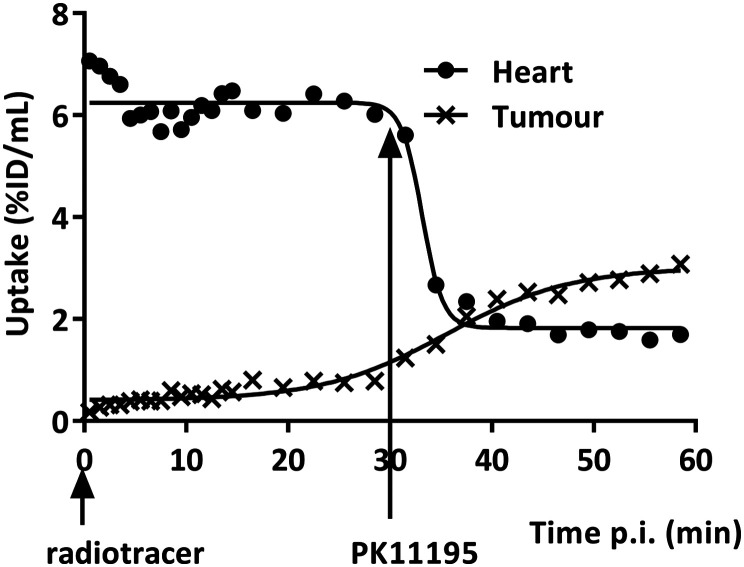
Radiopharmaceuticals that target the translocator protein 18 kDa (TSPO) have been investigated with positron emission tomography (PET) to study neuroinflammation, neurodegeneration and cancer. We have developed the novel, achiral, 2-phenylimidazo[1,2-a]pyridine, PBR316 that targets the translocator protein 18 kDa (TSPO) that addresses some of the limitations inherent in current TSPO ligands; namely specificity in binding, blood brain barrier permeability, metabolism and insensitivity to TSPO binding in subjects as a result of rs6971 polymorphism. PBR316 has high nanomolar affinity (4.7-6.0 nM) for the TSPO, >5000 nM for the central benzodiazepine receptor (CBR) and low sensitivity to rs6971 polymorphism with a low affinity binders (LABs) to high affinity binders (HABs) ratio of 1.5. [18F]PBR316 was prepared in 20 ± 5% radiochemical yield, >99% radiochemical purity and a molar activity of 160-400 GBq μmol-1. Biodistribution in rats showed high uptake of [18F]PBR316 in organs known to express TSPO such as heart (3.9%) and adrenal glands (7.5% ID per g) at 1 h. [18F]PBR316 entered the brain and accumulated in TSPO-expressing regions with an olfactory bulb to brain ratio of 3 at 15 min and 7 at 4 h. Radioactivity was blocked by PK11195 and Ro 5-4864 but not Flumazenil. Metabolite analysis showed that radioactivity in adrenal glands and the brain was predominantly due to the intact radiotracer. PET-CT studies in mouse-bearing prostate tumour xenografts indicated biodistribution similar to rats with radioactivity in the tumour increasing with time. [18F]PBR316 shows in vitro binding that is insensitive to human polymorphism and has specific and selective in vivo binding to the TSPO. [18F]PBR316 is suitable for further biological and clinical studies.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Preclinical evaluation of (S)-[18F]GE387, a novel 18-kDa translocator protein (TSPO) PET radioligand with low binding sensitivity to human polymorphism rs6971.Eur J Nucl Med Mol Imaging. 2021 Dec;49(1):125-136. doi: 10.1007/s00259-021-05495-w. Epub 2021 Aug 18. Eur J Nucl Med Mol Imaging. 2021. PMID: 34405276 Free PMC article.
-
Development and evaluation of [11C]DPA-813 and [18F]DPA-814: novel TSPO PET tracers insensitive to human single nucleotide polymorphism rs6971.Eur J Nucl Med Mol Imaging. 2025 Jun;52(7):2658-2670. doi: 10.1007/s00259-025-07109-1. Epub 2025 Feb 5. Eur J Nucl Med Mol Imaging. 2025. PMID: 39907797 Free PMC article.
-
Radiosynthesis and characterization of [18F]BS224: a next-generation TSPO PET ligand insensitive to the rs6971 polymorphism.Eur J Nucl Med Mol Imaging. 2021 Dec;49(1):110-124. doi: 10.1007/s00259-021-05617-4. Epub 2021 Nov 16. Eur J Nucl Med Mol Imaging. 2021. PMID: 34783879 Free PMC article.
-
Recent developments on PET radiotracers for TSPO and their applications in neuroimaging.Acta Pharm Sin B. 2021 Feb;11(2):373-393. doi: 10.1016/j.apsb.2020.08.006. Epub 2020 Aug 25. Acta Pharm Sin B. 2021. PMID: 33643818 Free PMC article. Review.
-
Positron emission tomography to image cerebral neuroinflammation in ischaemic stroke: a pilot study.Southampton (UK): NIHR Journals Library; 2020 Feb. Southampton (UK): NIHR Journals Library; 2020 Feb. PMID: 32023020 Free Books & Documents. Review.
Cited by
-
Translocator Protein 18 kDa (TSPO): A Promising Molecular Target for Image-Guided Surgery of Solid Cancers.Adv Pharm Bull. 2024 Mar;14(1):86-104. doi: 10.34172/apb.2024.015. Epub 2023 Oct 14. Adv Pharm Bull. 2024. PMID: 38585455 Free PMC article. Review.
-
TSPO Radioligands for Neuroinflammation: An Overview.Molecules. 2024 Sep 5;29(17):4212. doi: 10.3390/molecules29174212. Molecules. 2024. PMID: 39275061 Free PMC article. Review.
-
The 18-kDa Translocator Protein PET Tracers as a Diagnostic Marker for Neuroinflammation: Development and Current Standing.ACS Omega. 2022 Apr 18;7(17):14412-14429. doi: 10.1021/acsomega.2c00588. eCollection 2022 May 3. ACS Omega. 2022. PMID: 35557664 Free PMC article. Review.
-
18F-Radiolabeled Translocator Protein (TSPO) PET Tracers: Recent Development of TSPO Radioligands and Their Application to PET Study.Pharmaceutics. 2022 Nov 21;14(11):2545. doi: 10.3390/pharmaceutics14112545. Pharmaceutics. 2022. PMID: 36432736 Free PMC article. Review.
-
Imaging neuroinflammation with TSPO: A new perspective on the cellular sources and subcellular localization.Pharmacol Ther. 2022 Jun;234:108048. doi: 10.1016/j.pharmthera.2021.108048. Epub 2021 Nov 27. Pharmacol Ther. 2022. PMID: 34848203 Free PMC article. Review.
References
-
- Selvaraj V. Tu L. N. J. Endocrinol. 2016;231:R1–R30. - PubMed
-
- Papadopoulos V. Baraldi M. Guilarte T. R. Knudsen T. B. Lacapere J. J. Lindemann P. Norenberg M. D. Nutt D. Weizman A. Zhang M. R. Gavish M. Trends Pharmacol. Sci. 2006;27:402–409. - PubMed
-
- Han Z. Slack R. S. Li W. Papadopoulos V. J. Recept. Signal Transduction. 2003;23:225–238. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous