

Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations
- PMID: 34355210
- PMCID: PMC8336924
- DOI: 10.1016/j.crmeth.2021.100014
Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations
Abstract
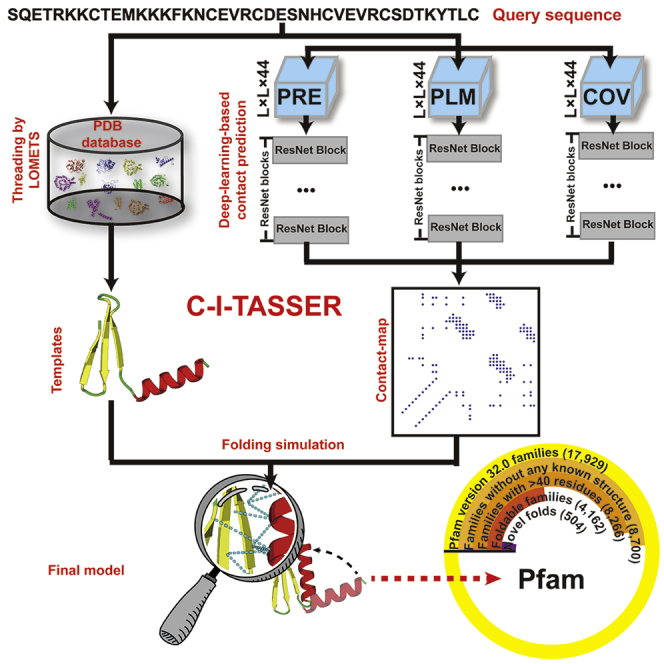
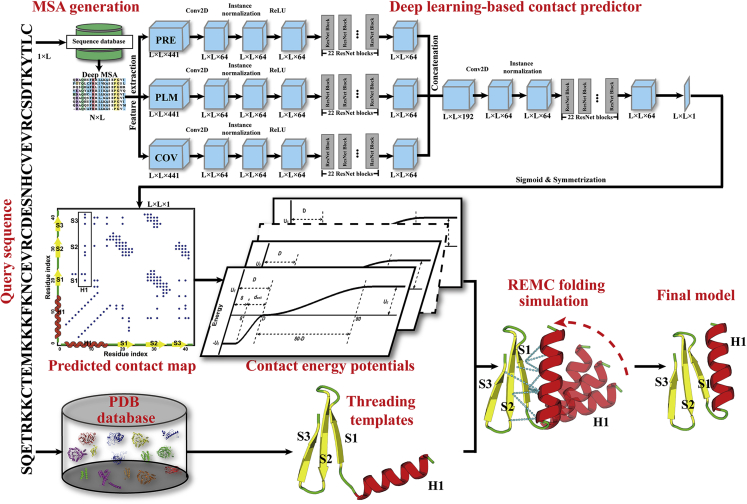
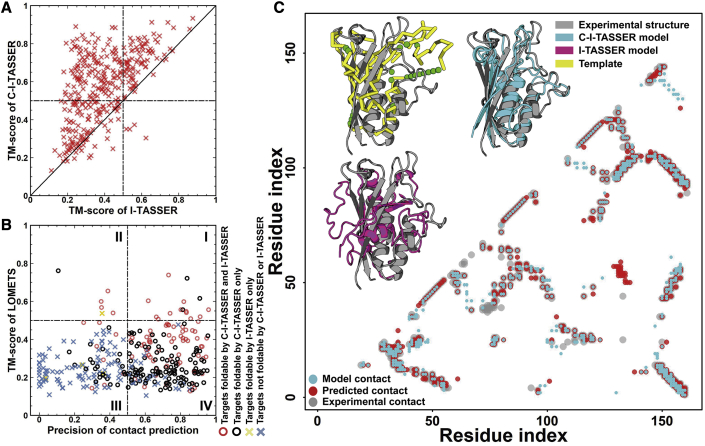
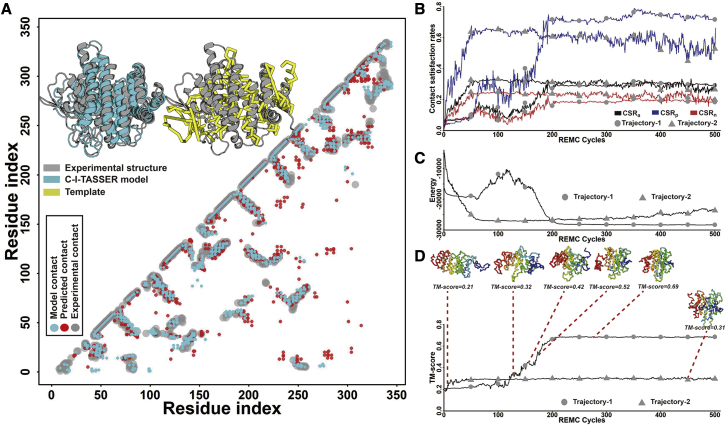
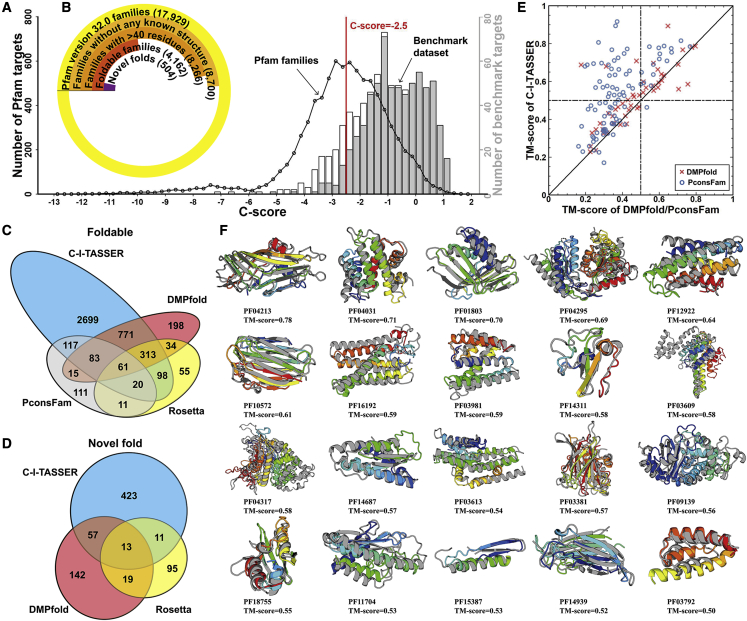
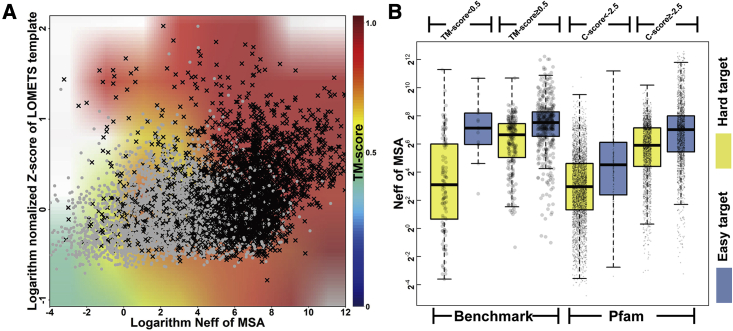
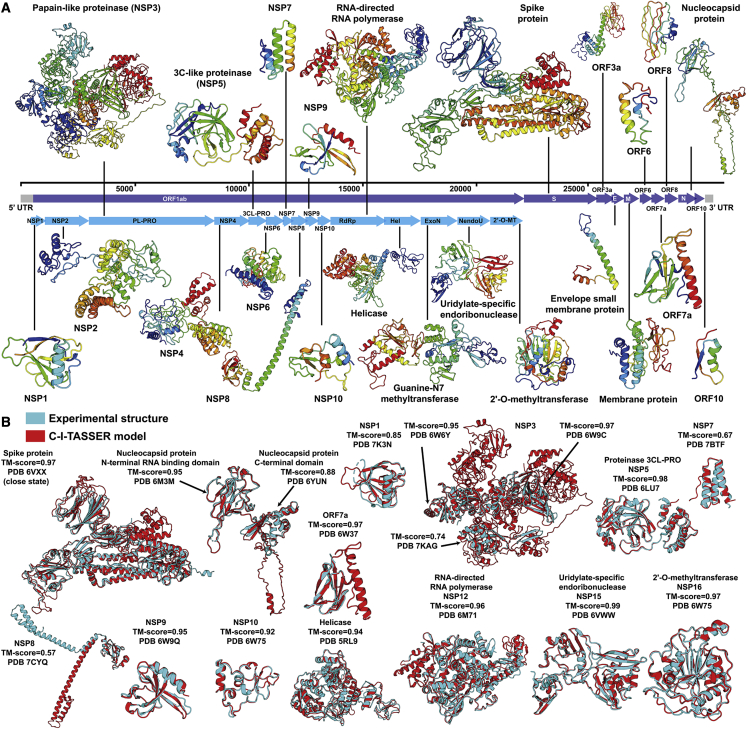
Structure prediction for proteins lacking homologous templates in the Protein Data Bank (PDB) remains a significant unsolved problem. We developed a protocol, C-I-TASSER, to integrate interresidue contact maps from deep neural-network learning with the cutting-edge I-TASSER fragment assembly simulations. Large-scale benchmark tests showed that C-I-TASSER can fold more than twice the number of non-homologous proteins than the I-TASSER, which does not use contacts. When applied to a folding experiment on 8,266 unsolved Pfam families, C-I-TASSER successfully folded 4,162 domain families, including 504 folds that are not found in the PDB. Furthermore, it created correct folds for 85% of proteins in the SARS-CoV-2 genome, despite the quick mutation rate of the virus and sparse sequence profiles. The results demonstrated the critical importance of coupling whole-genome and metagenome-based evolutionary information with optimal structure assembly simulations for solving the problem of non-homologous protein structure prediction.
Conflict of interest statement
DECLARATION OF INTERESTS The authors declare no competing interests.
Figures
References
-
- Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
