

Introduction to Single-Cell DNA Methylation Profiling Methods
- PMID: 34356635
- PMCID: PMC8301785
- DOI: 10.3390/biom11071013
Introduction to Single-Cell DNA Methylation Profiling Methods
Abstract
DNA methylation is an epigenetic mechanism that is related to mammalian cellular differentiation, gene expression regulation, and disease. In several studies, DNA methylation has been identified as an effective marker to identify differences between cells. In this review, we introduce single-cell DNA-methylation profiling methods, including experimental strategies and approaches to computational data analysis. Furthermore, the blind spots of the basic analysis and recent alternatives are briefly described. In addition, we introduce well-known applications and discuss future development.
Keywords: DNA methylation; bioinformatics; single cell.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
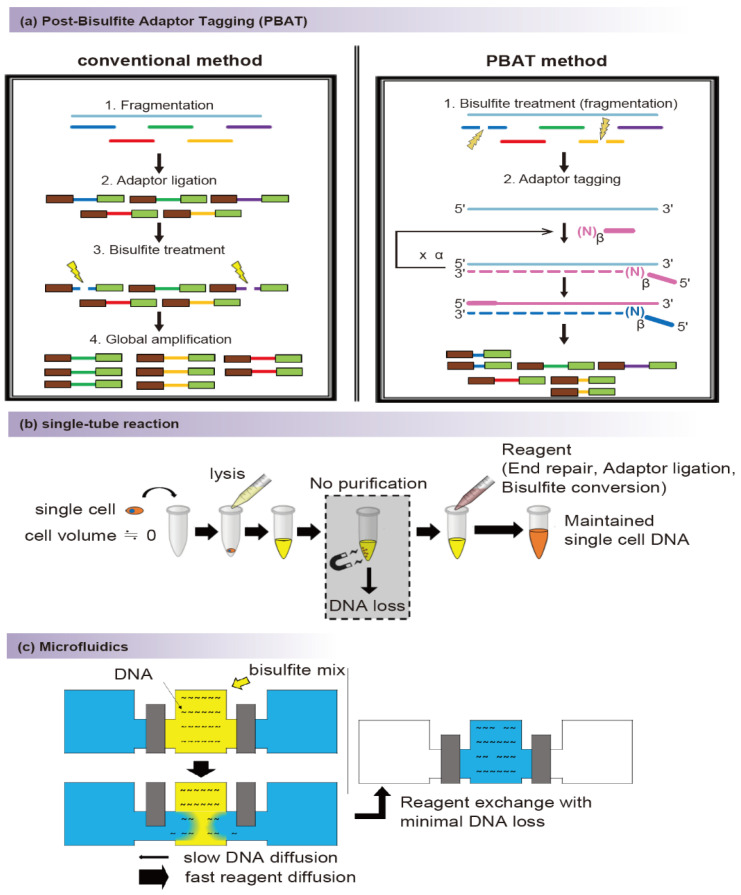
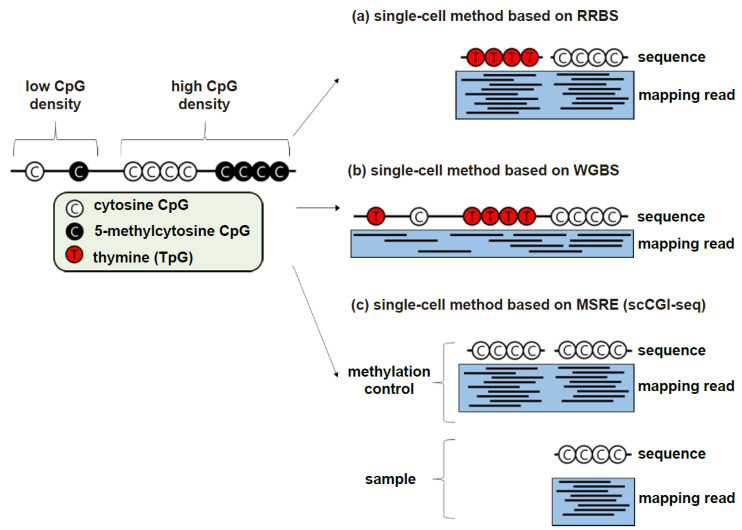
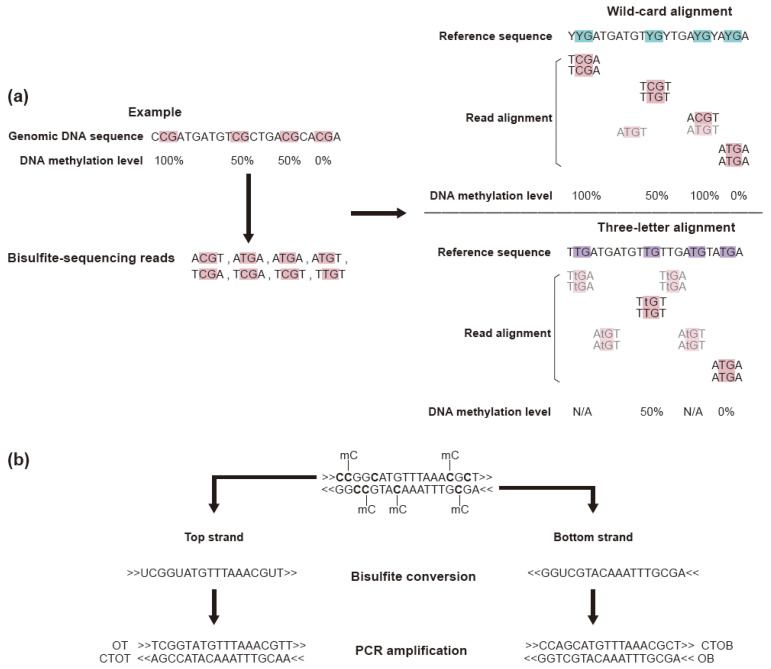
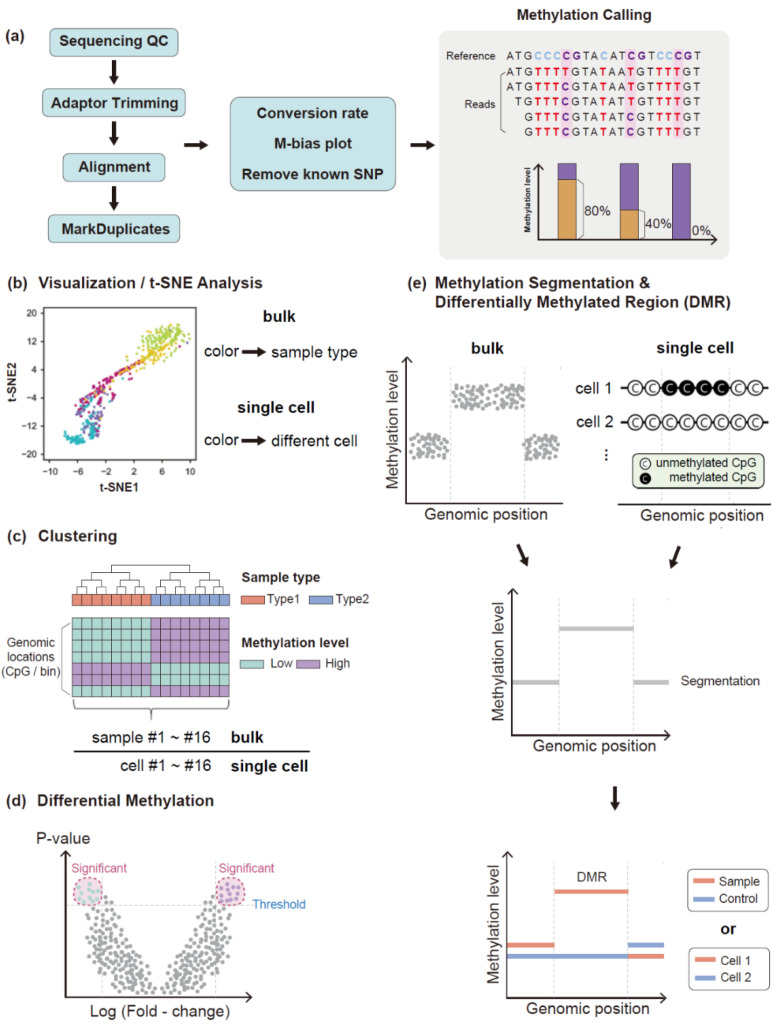
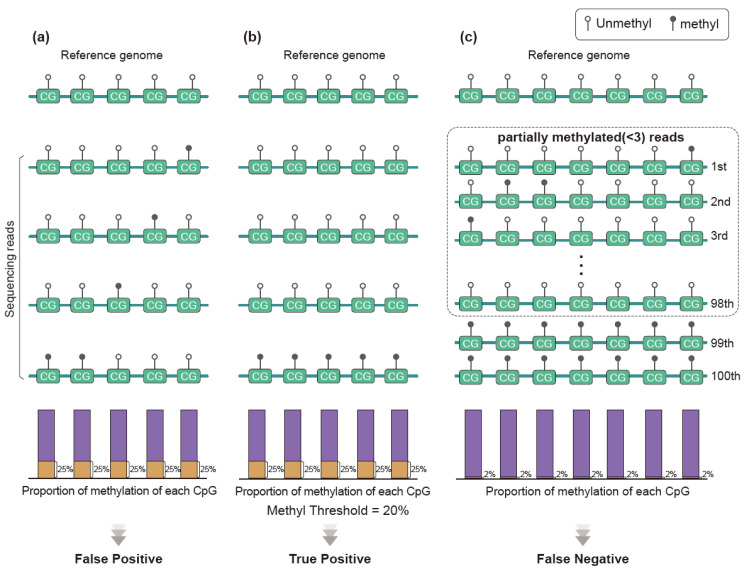
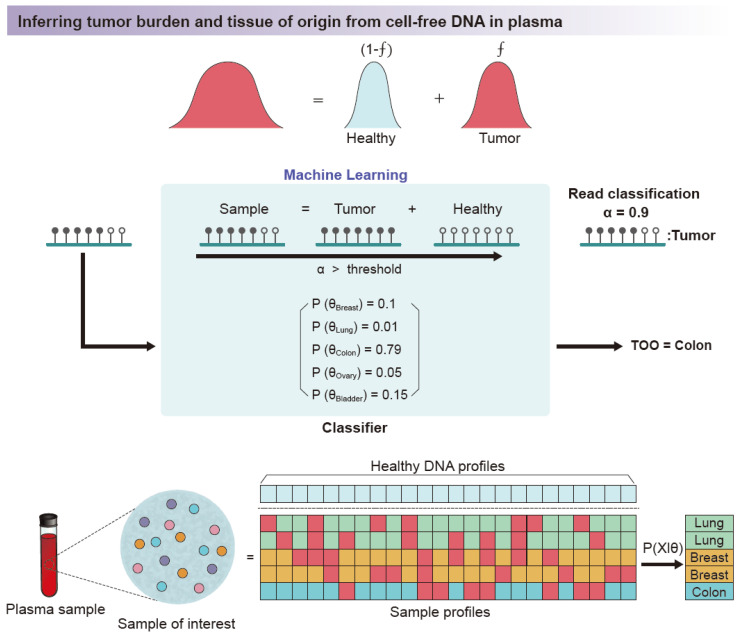
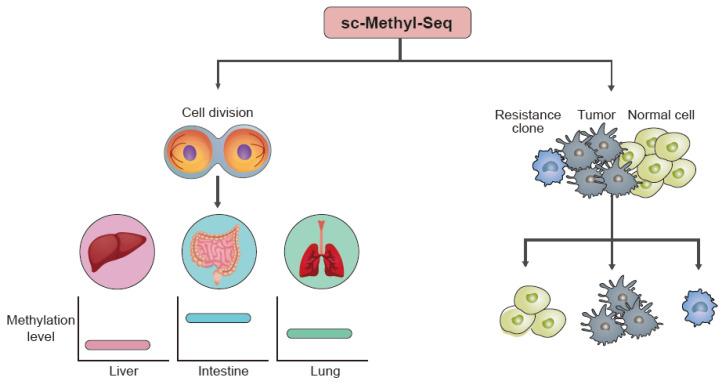
References
Publication types
MeSH terms
Grants and funding
- MSIT; 2016M3A9B6948494/Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government
- MSIT; 2018R1A2A1A05079172/Mid-career Researcher Program of the National Research Foundation (NRF) funded by the Korean government
- MSIT; 2021R1A2C2008490/National Research Foundation (NRF) funded by the Korean government
- HI18C2282, HI14C1277/Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea
LinkOut - more resources
Full Text Sources
Other Literature Sources
