

Elexacaftor-Tezacaftor-Ivacaftor Therapy for Cystic Fibrosis Patients with The F508del/Unknown Genotype
- PMID: 34356748
- PMCID: PMC8300667
- DOI: 10.3390/antibiotics10070828
Elexacaftor-Tezacaftor-Ivacaftor Therapy for Cystic Fibrosis Patients with The F508del/Unknown Genotype
Abstract
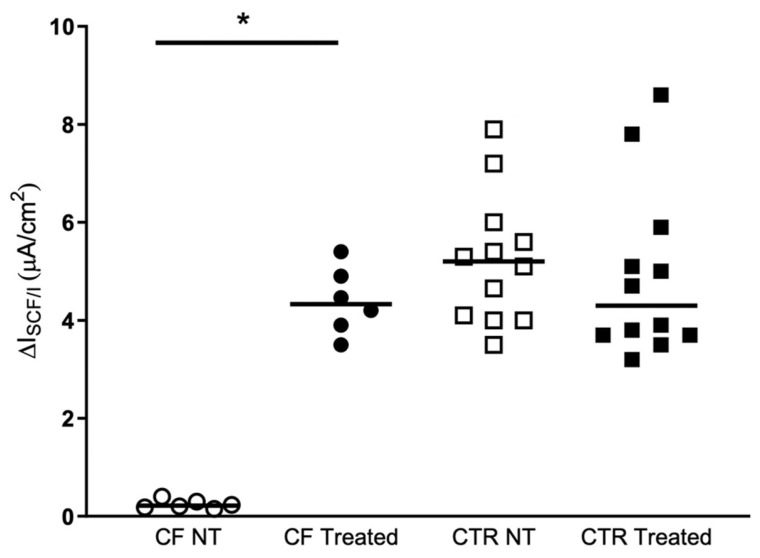
The new CFTR modulator combination, elexacaftor/tezacaftor/ivacaftor (Trikafta) was approved by the FDA in October 2019 for treatment of Cystic Fibrosis in patients 6 years of age or older who have at least one F508del mutation in one allele and a minimal-function or another F508del mutation in the other allele. However, there is a group of patients, in addition to those with rare mutations, in which despite the presence of a F508del in one allele, it was not possible to identify any mutation in the other allele. To date, these patients are excluded from treatment with Trikafta in Italy, where the CF patients carrying F508del/unknown represent about 1.3% (71 patients) of the overall Italian CF patients. In this paper we show that the Trikafta treatment of nasal epithelial cells, derived from F508del/Unknown patients, results in a significant rescue of CFTR activity. Based on our findings, we think that the F508del/Unknown patients considered in this study could obtain clinical benefits from Trikafta treatment, and we strongly suggest their eligibility for this type of treatment. This study, adding further evidence in the literature, once again confirms the validity of functional studies on nasal cells in the cystic fibrosis theratyping and personalized medicine.
Keywords: CFTR; cystic fibrosis; functional characterization; personalized medicine; rare mutation; theratyping.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Veit G., Avramescu R.G., Chiang A.N., Houck S.A., Cai Z., Peters K.W., Hong J.S., Pollard H.B., Guggino W.B., Balch W.E., et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell. 2016;27:424–433. doi: 10.1091/mbc.e14-04-0935. - DOI - PMC - PubMed
-
- Davies J.C., Cunningham S., Harris W.T., Lapey A., Regelmann W.E., Sawicki G.S., Southern K.W., Robertson S., Green Y., Cooke J., et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir. Med. 2016;4:107–115. doi: 10.1016/S2213-2600(15)00545-7. - DOI - PMC - PubMed
-
- Ren C.L., Morgan R.L., Oermann C., Resnick H.E., Brady C., Campbell A., DeNagel R., Guill M., Hoag J., Lipton A., et al. Cystic Fibrosis Foundation Pulmonary Guidelines. Use of Cystic Fibrosis Transmembrane Conductance Regulator Modulator Therapy in Patients with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2018;15:271–280. doi: 10.1513/AnnalsATS.201707-539OT. - DOI - PubMed
-
- Davies J.C., Wainwright C.E., Canny G.J., Chilvers M.A., Howenstine M.S., Munck A., Mainz J.G., Rodriguez S., Li H., Yen K., et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013;187:1219–1225. doi: 10.1164/rccm.201301-0153OC. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
