

PhyloFisher: A phylogenomic package for resolving eukaryotic relationships
- PMID: 34358228
- PMCID: PMC8345874
- DOI: 10.1371/journal.pbio.3001365
PhyloFisher: A phylogenomic package for resolving eukaryotic relationships
Abstract
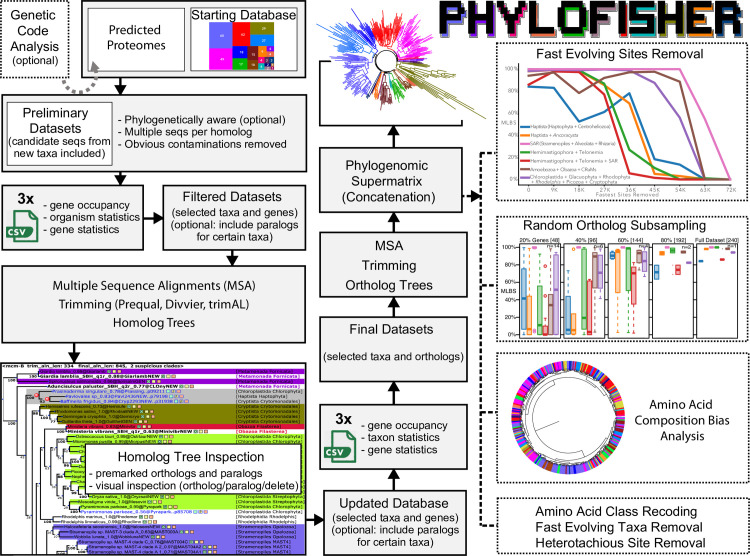
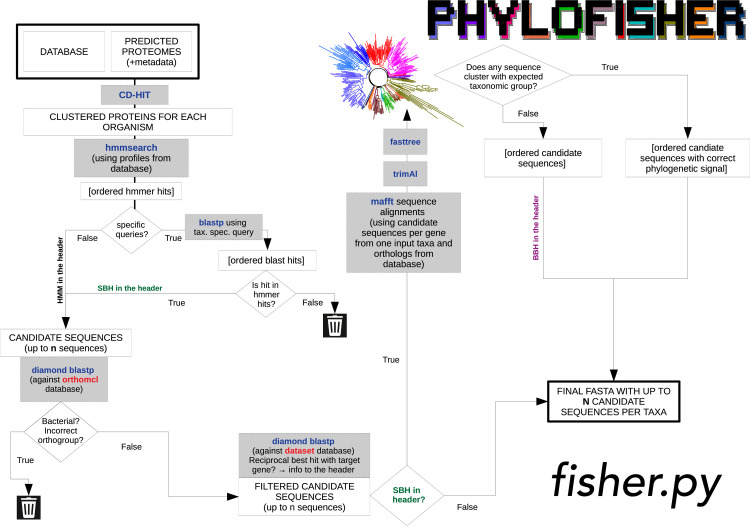
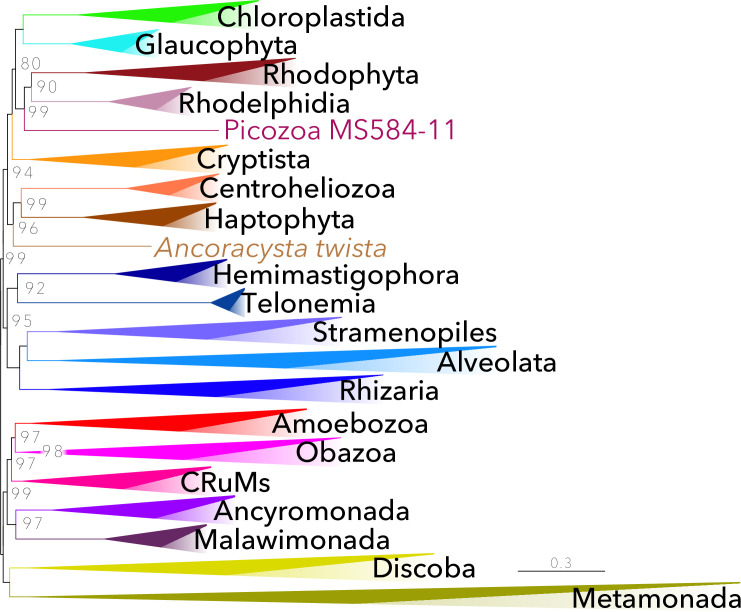
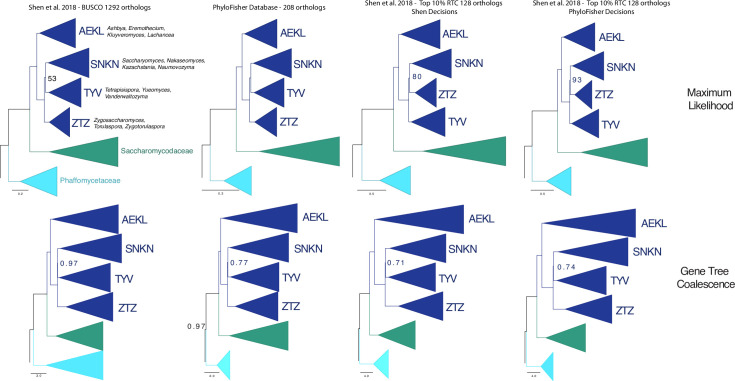
Phylogenomic analyses of hundreds of protein-coding genes aimed at resolving phylogenetic relationships is now a common practice. However, no software currently exists that includes tools for dataset construction and subsequent analysis with diverse validation strategies to assess robustness. Furthermore, there are no publicly available high-quality curated databases designed to assess deep (>100 million years) relationships in the tree of eukaryotes. To address these issues, we developed an easy-to-use software package, PhyloFisher (https://github.com/TheBrownLab/PhyloFisher), written in Python 3. PhyloFisher includes a manually curated database of 240 protein-coding genes from 304 eukaryotic taxa covering known eukaryotic diversity, a novel tool for ortholog selection, and utilities that will perform diverse analyses required by state-of-the-art phylogenomic investigations. Through phylogenetic reconstructions of the tree of eukaryotes and of the Saccharomycetaceae clade of budding yeasts, we demonstrate the utility of the PhyloFisher workflow and the provided starting database to address phylogenetic questions across a large range of evolutionary time points for diverse groups of organisms. We also demonstrate that undetected paralogy can remain in phylogenomic "single-copy orthogroup" datasets constructed using widely accepted methods such as all vs. all BLAST searches followed by Markov Cluster Algorithm (MCL) clustering and application of automated tree pruning algorithms. Finally, we show how the PhyloFisher workflow helps detect inadvertent paralog inclusions, allowing the user to make more informed decisions regarding orthology assignments, leading to a more accurate final dataset.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
