

Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond
- PMID: 34360549
- PMCID: PMC8346007
- DOI: 10.3390/ijms22157785
Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond
Abstract
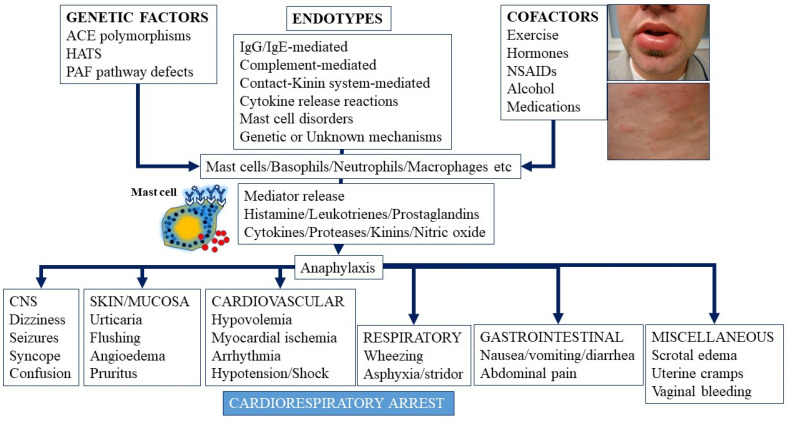
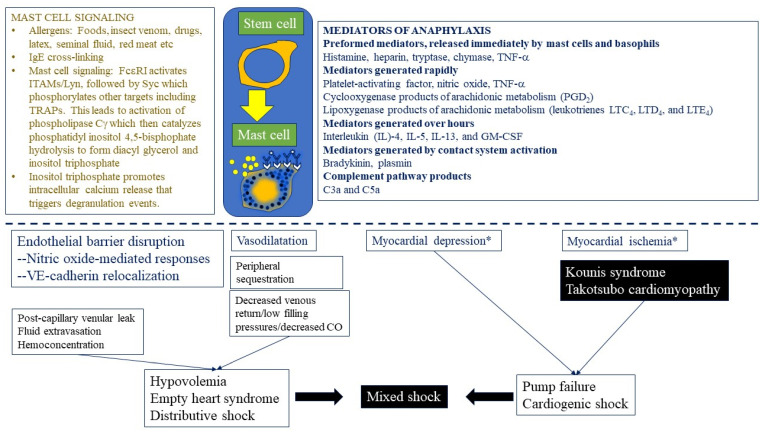
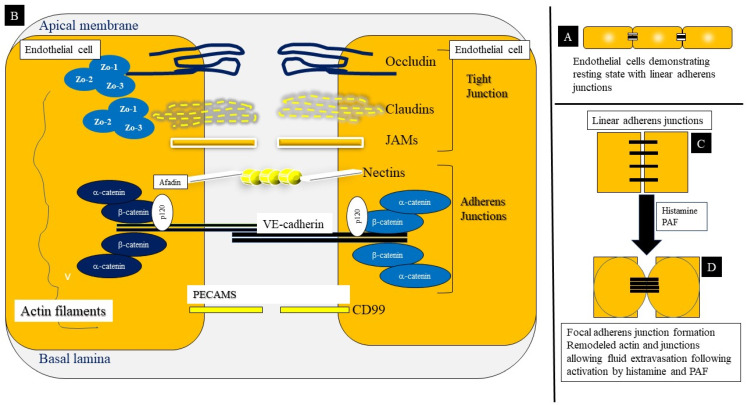
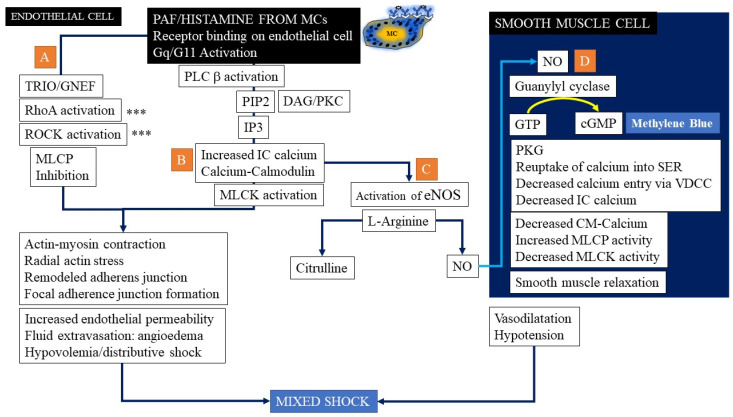
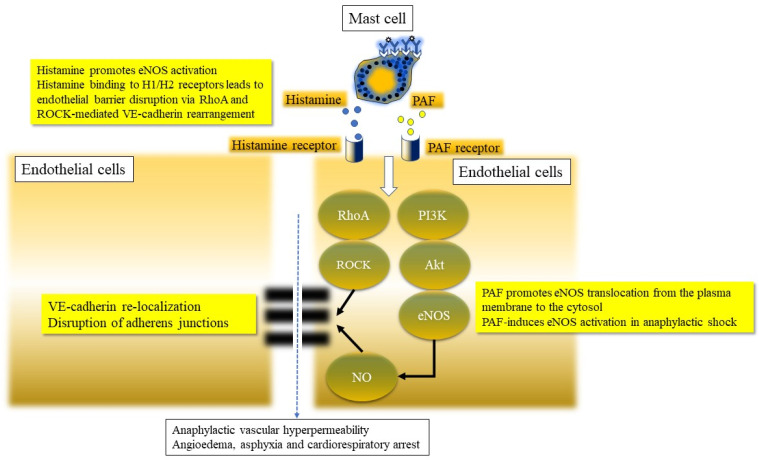
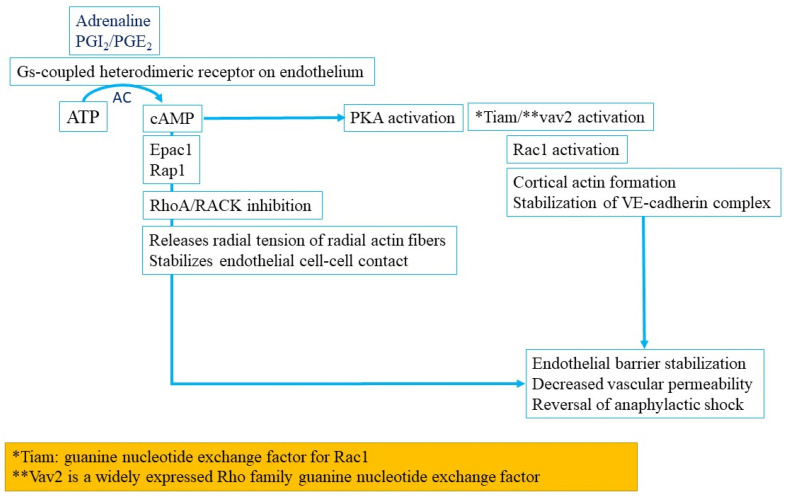
Anaphylaxis is a severe, acute, life-threatening multisystem allergic reaction resulting from the release of a plethora of mediators from mast cells culminating in serious respiratory, cardiovascular and mucocutaneous manifestations that can be fatal. Medications, foods, latex, exercise, hormones (progesterone), and clonal mast cell disorders may be responsible. More recently, novel syndromes such as delayed reactions to red meat and hereditary alpha tryptasemia have been described. Anaphylaxis manifests as sudden onset urticaria, pruritus, flushing, erythema, angioedema (lips, tongue, airways, periphery), myocardial dysfunction (hypovolemia, distributive or mixed shock and arrhythmias), rhinitis, wheezing and stridor. Vomiting, diarrhea, scrotal edema, uterine cramps, vaginal bleeding, urinary incontinence, dizziness, seizures, confusion, and syncope may occur. The traditional (or classical) pathway is mediated via T cells, Th2 cytokines (such as IL-4 and 5), B cell production of IgE and subsequent crosslinking of the high affinity IgE receptor (FcεRI) on mast cells and basophils by IgE-antigen complexes, culminating in mast cell and basophil degranulation. Degranulation results in the release of preformed mediators (histamine, heparin, tryptase, chymase, carboxypeptidase, cathepsin G and tumor necrosis factor alpha (TNF-α), and of de novo synthesized ones such as lipid mediators (cysteinyl leukotrienes), platelet activating factor (PAF), cytokines and growth factors such as vascular endothelial growth factor (VEGF). Of these, histamine, tryptase, cathepsin G, TNF-α, LTC4, PAF and VEGF can increase vascular permeability. Recent data suggest that mast cell-derived histamine and PAF can activate nitric oxide production from endothelium and set into motion a signaling cascade that leads to dilatation of blood vessels and dysfunction of the endothelial barrier. The latter, characterized by the opening of adherens junctions, leads to increased capillary permeability and fluid extravasation. These changes contribute to airway edema, hypovolemia, and distributive shock, with potentially fatal consequences. In this review, besides mechanisms (endotypes) underlying IgE-mediated anaphylaxis, we also provide a brief overview of IgG-, complement-, contact system-, cytokine- and mast cell-mediated reactions that can result in phenotypes resembling IgE-mediated anaphylaxis. Such classifications can lead the way to precision medicine approaches to the management of this complex disease.
Keywords: allergic reaction; allergy; anaphylactic shock; anaphylaxis; angioedema; coagulation; complement; cytokines; epinephrine; food allergy; histamine; hypotension; mast cell; tryptase.
Conflict of interest statement
The authors declare no conflict of interests.
Figures


References
-
- Sampson H.A., Muñoz-Furlong A., Campbell R.L., Adkinson N.F., Bock S.A., Branum A., Brown S., Camargo C.A., Cydulka R., Galli S.J., et al. Second symposium on the definition and management of anaphylaxis: Summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J. Allergy Clin. Immunol. 2006;117:391–397. doi: 10.1016/j.jaci.2005.12.1303. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
