

Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development
- PMID: 34360669
- PMCID: PMC8348669
- DOI: 10.3390/ijms22157896
Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development
Abstract
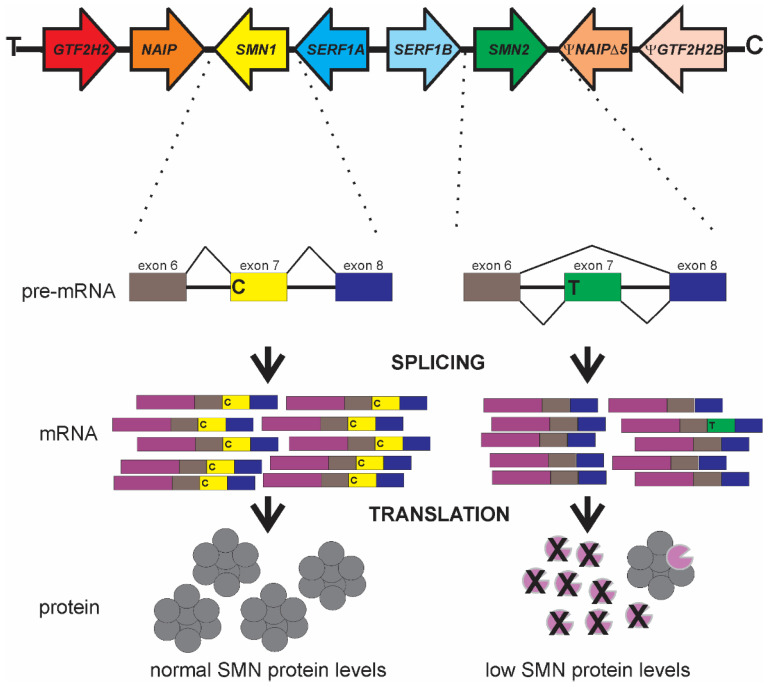
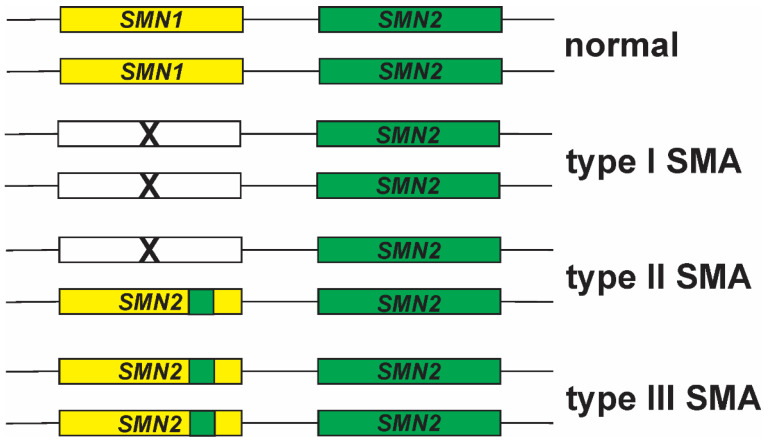
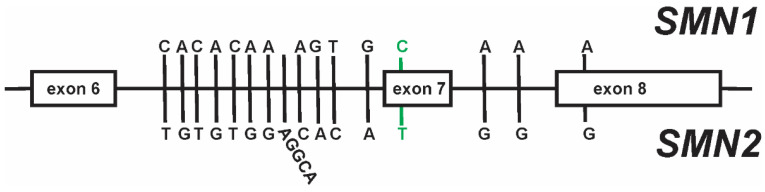
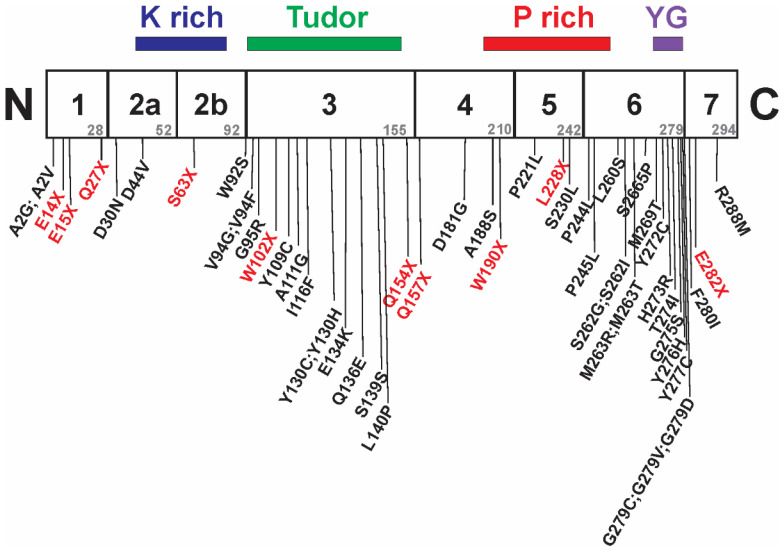
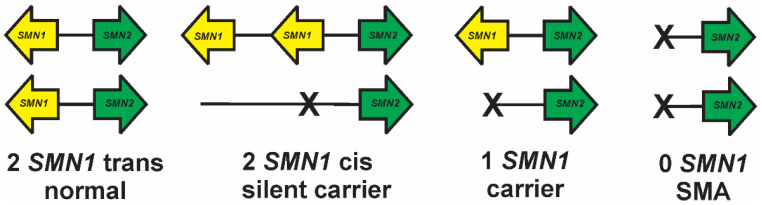
Spinal muscular atrophy (SMA) is a leading genetic cause of infant death worldwide that is characterized by loss of spinal motor neurons leading to muscle weakness and atrophy. SMA results from the loss of survival motor neuron 1 (SMN1) gene but retention of its paralog SMN2. The copy numbers of SMN1 and SMN2 are variable within the human population with SMN2 copy number inversely correlating with SMA severity. Current therapeutic options for SMA focus on increasing SMN2 expression and alternative splicing so as to increase the amount of SMN protein. Recent work has demonstrated that not all SMN2, or SMN1, genes are equivalent and there is a high degree of genomic heterogeneity with respect to the SMN genes. Because SMA is now an actionable disease with SMN2 being the primary target, it is imperative to have a comprehensive understanding of this genomic heterogeneity with respect to hybrid SMN1-SMN2 genes generated by gene conversion events as well as partial deletions of the SMN genes. This review will describe this genetic heterogeneity in SMA and its impact on disease phenotype as well as therapeutic efficacy.
Keywords: SMN1; SMN2; copy number variation; gene conversion; hybrid gene; modifier gene; precision medicine; spinal muscular atrophy; therapeutics.
Conflict of interest statement
The author declares no conflict of interest. The funders had no role in the writing of the manuscript or in the decision to publish the review.
Figures
References
-
- Sugarman E.A., Nagan N., Zhu H., Akmaev V.R., Zhou Z., Rohlfs A.M., Flynn K., Hendrickson B.C., Scholl T., Sirko-Osadsa D.A., et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of > 72400 specimens. Eur. J. Hum. Genet. 2012;20:27–32. doi: 10.1038/ejhg.2011.134. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
