

Recombination and lineage-specific mutations linked to the emergence of SARS-CoV-2
- PMID: 34362430
- PMCID: PMC8343217
- DOI: 10.1186/s13073-021-00943-6
Recombination and lineage-specific mutations linked to the emergence of SARS-CoV-2
Abstract
Background: The emergence of SARS-CoV-2 underscores the need to better understand the evolutionary processes that drive the emergence and adaptation of zoonotic viruses in humans. In the betacoronavirus genus, which also includes SARS-CoV and MERS-CoV, recombination frequently encompasses the receptor binding domain (RBD) of the Spike protein, which is responsible for viral binding to host cell receptors. In this work, we reconstruct the evolutionary events that have accompanied the emergence of SARS-CoV-2, with a special emphasis on the RBD and its adaptation for binding to its receptor, human ACE2.
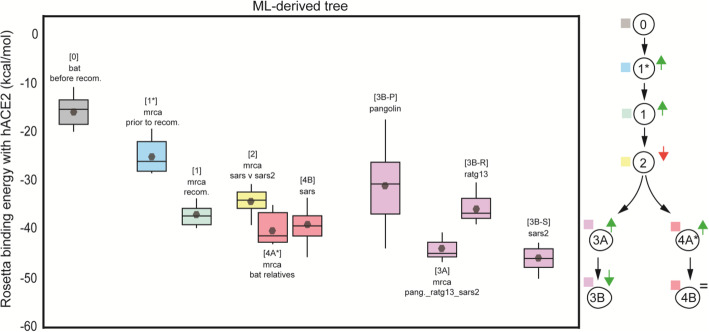
Methods: By means of phylogenetic and recombination analyses, we found evidence of a recombination event in the RBD involving ancestral linages to both SARS-CoV and SARS-CoV-2. We then assessed the effect of this recombination at protein level by reconstructing the RBD of the closest ancestors to SARS-CoV-2, SARS-CoV, and other Sarbecoviruses, including the most recent common ancestor of the recombining clade. The resulting information was used to measure and compare, in silico, their ACE2-binding affinities using the physics-based trRosetta algorithm.
Results: We show that, through an ancestral recombination event, SARS-CoV and SARS-CoV-2 share an RBD sequence that includes two insertions (positions 432-436 and 460-472), as well as the variants 427N and 436Y. Both 427N and 436Y belong to a helix that interacts directly with the human ACE2 (hACE2) receptor. Reconstruction of ancestral states, combined with protein-binding affinity analyses, suggests that the recombination event involving ancestral strains of SARS-CoV and SARS-CoV-2 led to an increased affinity for hACE2 binding and that alleles 427N and 436Y significantly enhanced affinity as well.
Conclusions: We report an ancestral recombination event affecting the RBD of both SARS-CoV and SARS-CoV-2 that was associated with an increased binding affinity to hACE2. Structural modeling indicates that ancestors of SARS-CoV-2 may have acquired the ability to infect humans decades ago. The binding affinity with the human receptor would have been subsequently boosted in SARS-CoV and SARS-CoV-2 through further mutations in RBD.
Keywords: Receptor binding affinity; Recombination; SARS-CoV-2; Zoonosis.
© 2021. The Author(s).
Conflict of interest statement
R.R. is a member of the SAB of AimedBio, consults for Arquimea Research and is founder of Genotwin. PKS is a member of the SAB or Board of Directors of Applied Biomath LLC, Glencoe Software Inc., and RareCyte Inc. and has equity in these companies; his is also on the SAB of NanoString Inc. In the last 5 years, the Sorger lab has received research funding from Novartis and Merck. Sorger declares that none of these relationships are directly or indirectly related to the content of this manuscript. The remaining authors declare that they have no competing interests.
Figures





Update of
-
Recombination and lineage-specific mutations linked to the emergence of SARS-CoV-2.bioRxiv [Preprint]. 2021 Apr 6:2020.02.10.942748. doi: 10.1101/2020.02.10.942748. bioRxiv. 2021. Update in: Genome Med. 2021 Aug 6;13(1):124. doi: 10.1186/s13073-021-00943-6. PMID: 32511304 Free PMC article. Updated. Preprint.
References
-
- W.H.O . Coronavirus disease 2019 (COVID-19) Situation Report - 22 June. 2021.
-
- Peng Zhou X-LY, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao K, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan F-X, Wang Y-Y, Xiao G, Shi Z-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(579):270–273. doi: 10.1038/s41586-020-2012-7. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
