

NLRP3 Inflammasome: A Starring Role in Amyloid-β- and Tau-Driven Pathological Events in Alzheimer's Disease
- PMID: 34366341
- PMCID: PMC8543248
- DOI: 10.3233/JAD-210268
NLRP3 Inflammasome: A Starring Role in Amyloid-β- and Tau-Driven Pathological Events in Alzheimer's Disease
Abstract
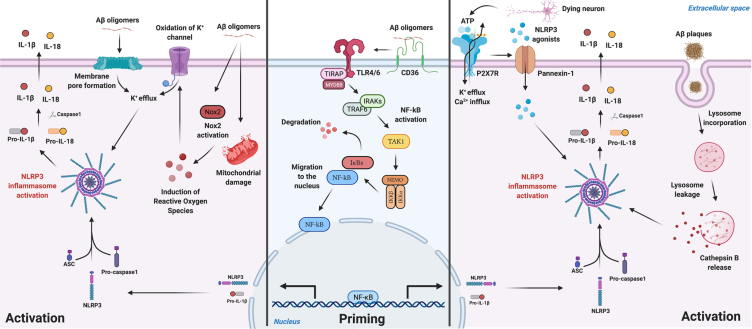
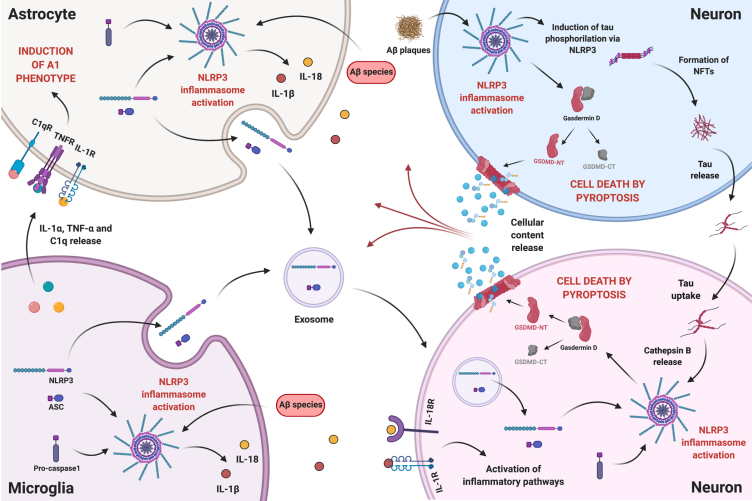
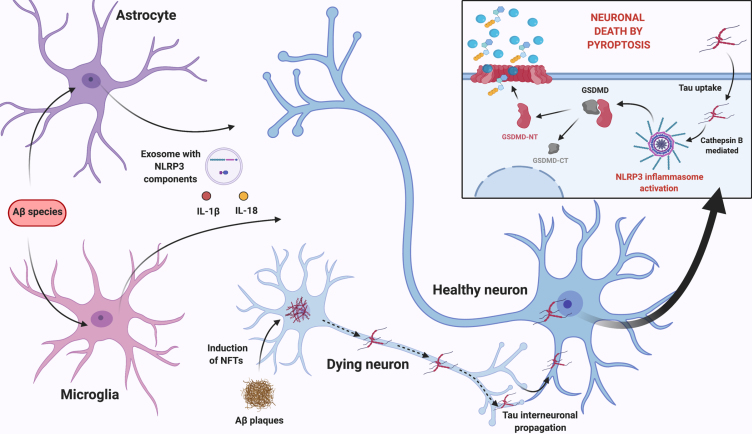
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease commonly diagnosed among the elderly population. AD is characterized by the loss of synaptic connections, neuronal death, and progressive cognitive impairment, attributed to the extracellular accumulation of senile plaques, composed by insoluble aggregates of amyloid-β (Aβ) peptides, and to the intraneuronal formation of neurofibrillary tangles shaped by hyperphosphorylated filaments of the microtubule-associated protein tau. However, evidence showed that chronic inflammatory responses, with long-lasting exacerbated release of proinflammatory cytokines by reactive glial cells, contribute to the pathophysiology of the disease. NLRP3 inflammasome (NLRP3), a cytosolic multiprotein complex sensor of a wide range of stimuli, was implicated in multiple neurological diseases, including AD. Herein, we review the most recent findings regarding the involvement of NLRP3 in the pathogenesis of AD. We address the mechanisms of NLRP3 priming and activation in glial cells by Aβ species and the potential role of neurofibrillary tangles and extracellular vesicles in disease progression. Neuronal death by NLRP3-mediated pyroptosis, driven by the interneuronal tau propagation, is also discussed. We present considerable evidence to claim that NLRP3 inhibition, is undoubtfully a potential therapeutic strategy for AD.
Keywords: Alzheimer’s disease; MCC950; NLRP3; amyloid-β; astrocytes; inflammasome; microglia; neuroinflammation; pro-inflammatory cytokines; pyroptosis; tau.
Conflict of interest statement
Authors’ disclosures available online (
Figures
References
-
- Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL (2015) Alzheimer’s disease. Nat Rev Dis Prim 1, 15056. - PubMed
-
- Stelzmann RA, Norman Schnitzlein H, Reed Murtagh F (1995) An English translation of Alzheimer’s 1907 paper, “über eine eigenartige erkankung der hirnrinde.”. Clin Anat 8, 429–431. - PubMed
-
- Querfurth HW, Laferla FM (2010) Alzheimer’s disease. N Engl J Med 9, 329–344. - PubMed
-
- Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12, 383–388. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
