

Crosstalk Between Dysfunctional Mitochondria and Inflammation in Glaucomatous Neurodegeneration
- PMID: 34366851
- PMCID: PMC8334009
- DOI: 10.3389/fphar.2021.699623
Crosstalk Between Dysfunctional Mitochondria and Inflammation in Glaucomatous Neurodegeneration
Abstract
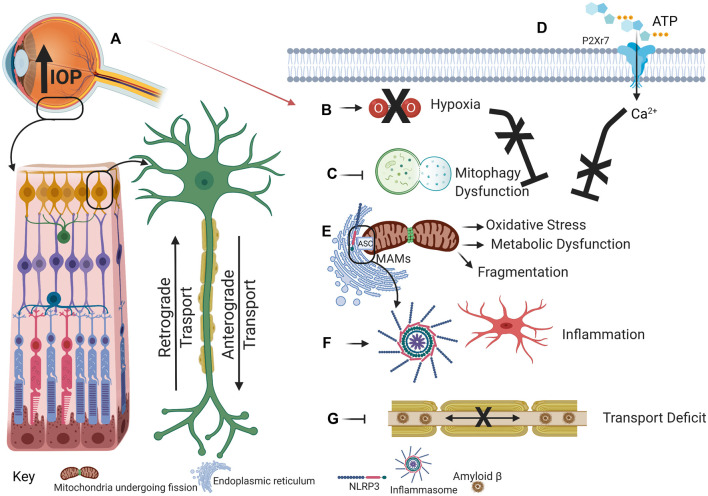
Mitochondrial dysfunction and excessive inflammatory responses are both sufficient to induce pathology in age-dependent neurodegenerations. However, emerging evidence indicates crosstalk between damaged mitochondrial and inflammatory signaling can exacerbate issues in chronic neurodegenerations. This review discusses evidence for the interaction between mitochondrial damage and inflammation, with a focus on glaucomatous neurodegeneration, and proposes that positive feedback resulting from this crosstalk drives pathology. Mitochondrial dysfunction exacerbates inflammatory signaling in multiple ways. Damaged mitochondrial DNA is a damage-associated molecular pattern, which activates the NLRP3 inflammasome; priming and activation of the NLRP3 inflammasome, and the resulting liberation of IL-1β and IL-18 via the gasdermin D pore, is a major pathway to enhance inflammatory responses. The rise in reactive oxygen species induced by mitochondrial damage also activates inflammatory pathways, while blockage of Complex enzymes is sufficient to increase inflammatory signaling. Impaired mitophagy contributes to inflammation as the inability to turnover mitochondria in a timely manner increases levels of ROS and damaged mtDNA, with the latter likely to stimulate the cGAS-STING pathway to increase interferon signaling. Mitochondrial associated ER membrane contacts and the mitochondria-associated adaptor molecule MAVS can activate NLRP3 inflammasome signaling. In addition to dysfunctional mitochondria increasing inflammation, the corollary also occurs, with inflammation reducing mitochondrial function and ATP production; the resulting downward spiral accelerates degeneration. Evidence from several preclinical models including the DBA/2J mouse, microbead injection and transient elevation of IOP, in addition to patient data, implicates both mitochondrial damage and inflammation in glaucomatous neurodegeneration. The pressure-dependent hypoxia and the resulting metabolic vulnerability is associated with mitochondrial damage and IL-1β release. Links between mitochondrial dysfunction and inflammation can occur in retinal ganglion cells, microglia cells and astrocytes. In summary, crosstalk between damaged mitochondria and increased inflammatory signaling enhances pathology in glaucomatous neurodegeneration, with implications for other complex age-dependent neurodegenerations like Alzheimer's and Parkinson's disease.
Keywords: NLRP3 infammasome; astrocyte; glaucoma; metabolic vulnerability; microglia; mitophagy; retinal ganglion cells.
Copyright © 2021 Jassim, Inman and Mitchell.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous