

Gene Expression Meta-Analysis Reveals Interferon-Induced Genes Associated With SARS Infection in Lungs
- PMID: 34367154
- PMCID: PMC8342995
- DOI: 10.3389/fimmu.2021.694355
Gene Expression Meta-Analysis Reveals Interferon-Induced Genes Associated With SARS Infection in Lungs
Abstract
Background: Severe Acute Respiratory Syndrome (SARS) corona virus (CoV) infections are a serious public health threat because of their pandemic-causing potential. This work is the first to analyze mRNA expression data from SARS infections through meta-analysis of gene signatures, possibly identifying therapeutic targets associated with major SARS infections.
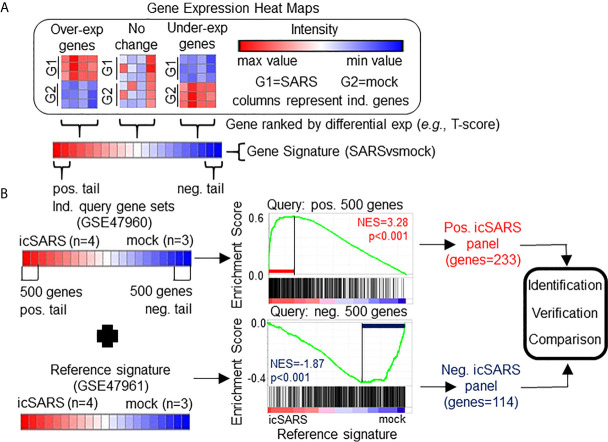
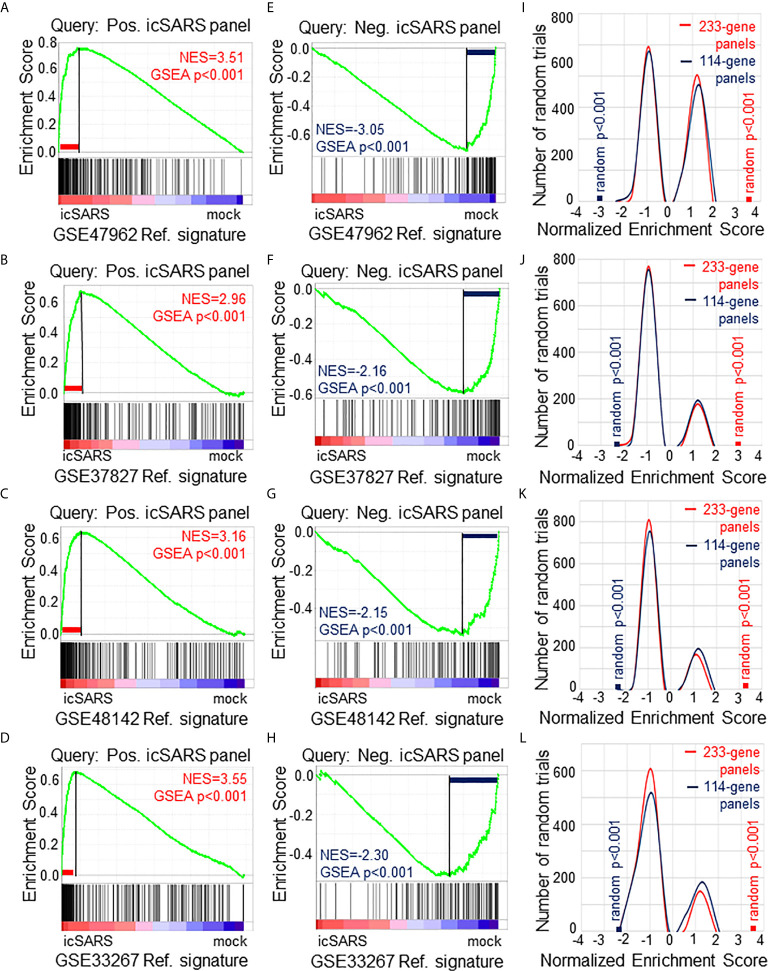
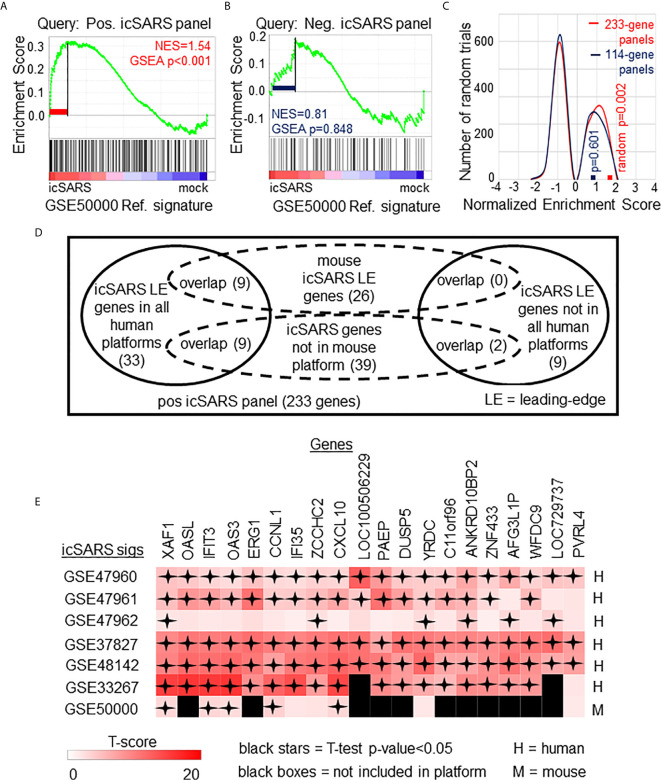
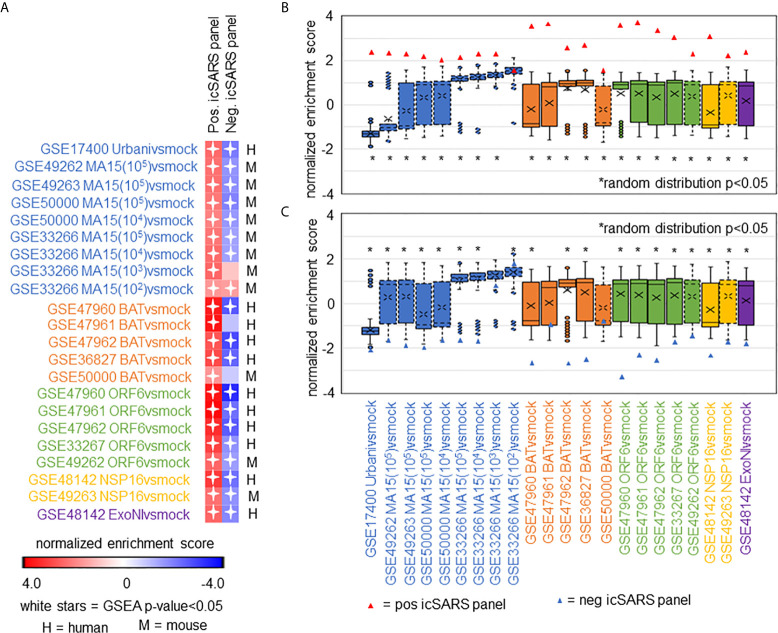
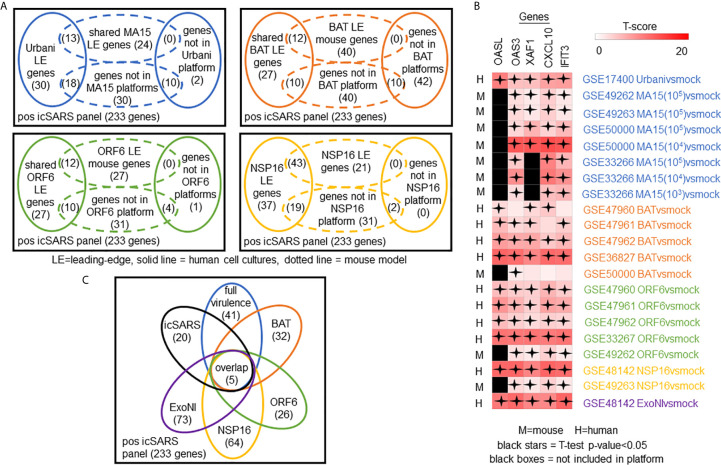
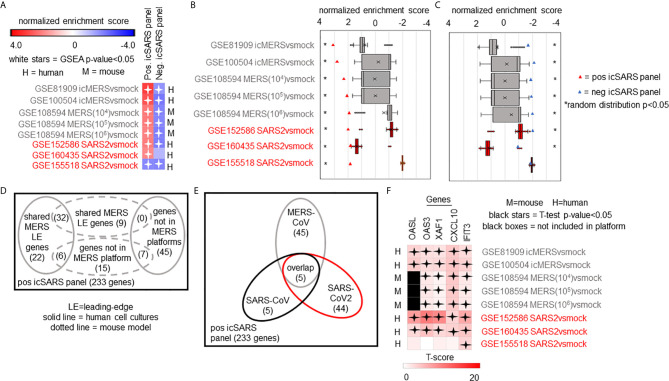
Methods: This work defines 37 gene signatures representing SARS-CoV, Middle East Respiratory Syndrome (MERS)-CoV, and SARS-CoV2 infections in human lung cultures and/or mouse lung cultures or samples and compares them through Gene Set Enrichment Analysis (GSEA). To do this, positive and negative infectious clone SARS (icSARS) gene panels are defined from GSEA-identified leading-edge genes between two icSARS-CoV derived signatures, both from human cultures. GSEA then is used to assess enrichment and identify leading-edge icSARS panel genes between icSARS gene panels and 27 other SARS-CoV gene signatures. The meta-analysis is expanded to include five MERS-CoV and three SARS-CoV2 gene signatures. Genes associated with SARS infection are predicted by examining the intersecting membership of GSEA-identified leading-edges across gene signatures.
Results: Significant enrichment (GSEA p<0.001) is observed between two icSARS-CoV derived signatures, and those leading-edge genes defined the positive (233 genes) and negative (114 genes) icSARS panels. Non-random significant enrichment (null distribution p<0.001) is observed between icSARS panels and all verification icSARSvsmock signatures derived from human cultures, from which 51 over- and 22 under-expressed genes are shared across leading-edges with 10 over-expressed genes already associated with icSARS infection. For the icSARSvsmock mouse signature, significant, non-random significant enrichment held for only the positive icSARS panel, from which nine genes are shared with icSARS infection in human cultures. Considering other SARS strains, significant, non-random enrichment (p<0.05) is observed across signatures derived from other SARS strains for the positive icSARS panel. Five positive icSARS panel genes, CXCL10, OAS3, OASL, IFIT3, and XAF1, are found across mice and human signatures regardless of SARS strains.
Conclusion: The GSEA-based meta-analysis approach used here identifies genes with and without reported associations with SARS-CoV infections, highlighting this approach's predictability and usefulness in identifying genes that have potential as therapeutic targets to preclude or overcome SARS infections.
Keywords: MERS-CoV; SARS-CoV; SARS-CoV2; coronavirus; gene expression; gene set enrichment analysis; meta-analysis.
Copyright © 2021 Park and Harris.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Li X, Luk HKH, Lau SKP, Woo PCY. Human Coronaviruses: General Features. Reference Module Biomed Sci (2019) 2019:B978–970-912-801238-801233.895704-801230. 10.1016/B978-0-12-801238-3.95704-0 - DOI
-
- Yount B, Roberts RS, Sims AC, Deming D, Frieman MB, Sparks J, et al. Severe Acute Respiratory Syndrome Coronavirus Group-Specific Open Reading Frames Encode Nonessential Functions for Replication in Cell Cultures and Mice. J Virol (2005) 79(23):14909–22. 10.1128/JVI.79.23.14909-14922.2005 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous