

Cell Fate Reprogramming in the Era of Cancer Immunotherapy
- PMID: 34367185
- PMCID: PMC8336566
- DOI: 10.3389/fimmu.2021.714822
Cell Fate Reprogramming in the Era of Cancer Immunotherapy
Abstract
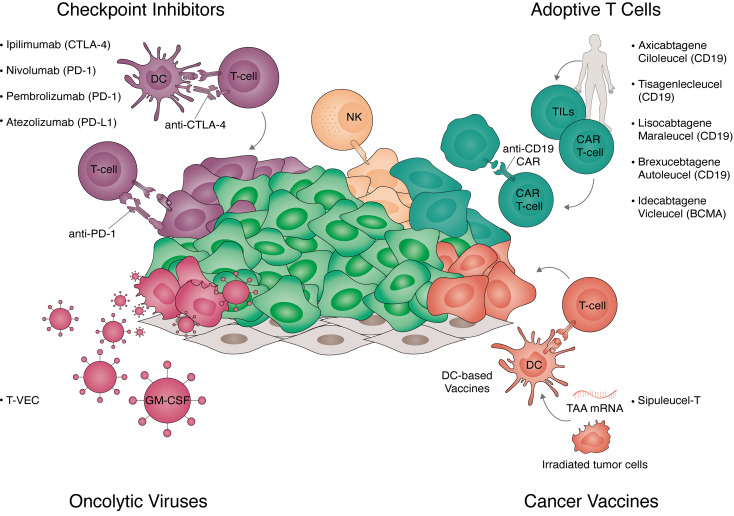
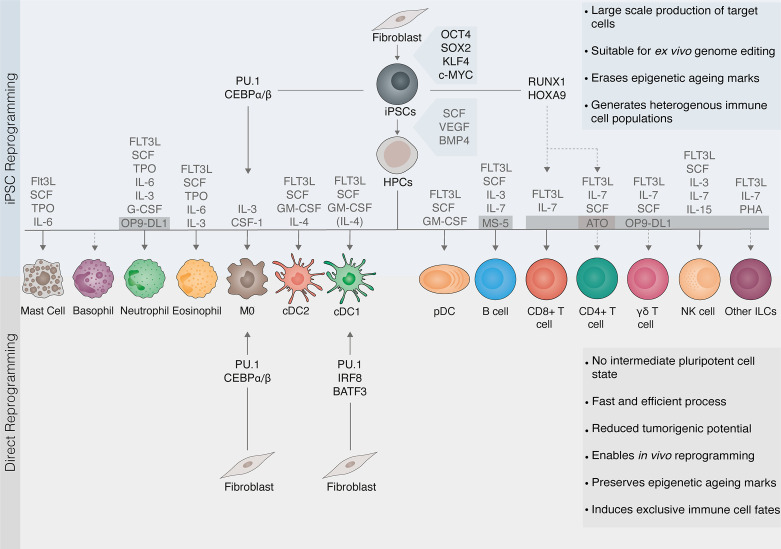
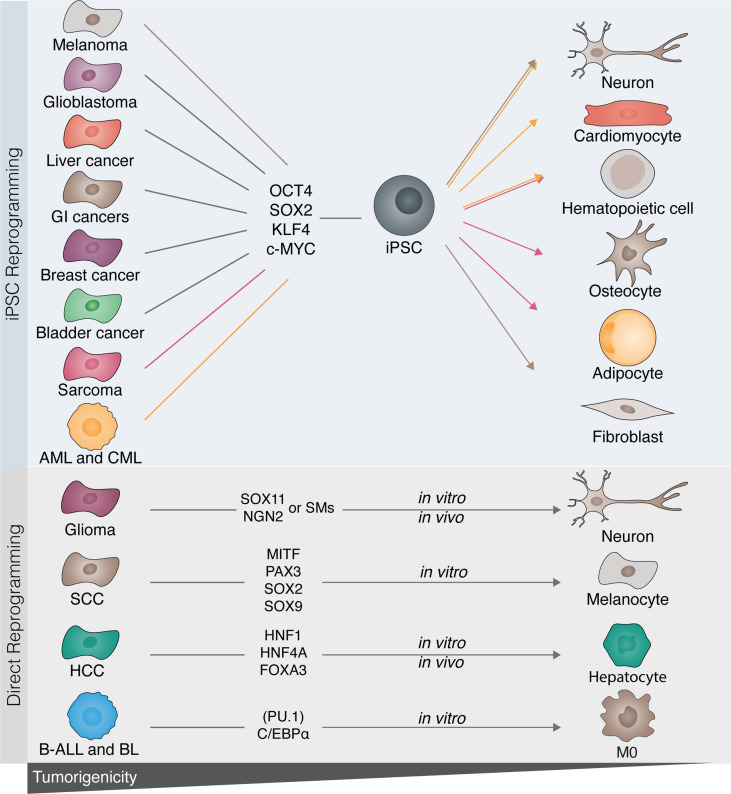
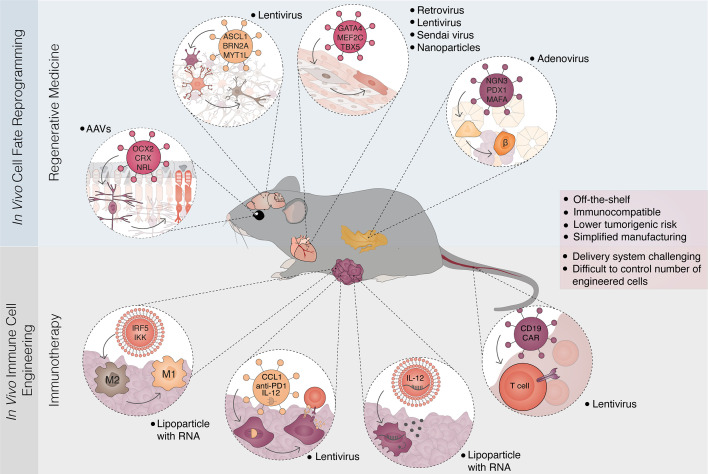
Advances in understanding how cancer cells interact with the immune system allowed the development of immunotherapeutic strategies, harnessing patients' immune system to fight cancer. Dendritic cell-based vaccines are being explored to reactivate anti-tumor adaptive immunity. Immune checkpoint inhibitors and chimeric antigen receptor T-cells (CAR T) were however the main approaches that catapulted the therapeutic success of immunotherapy. Despite their success across a broad range of human cancers, many challenges remain for basic understanding and clinical progress as only a minority of patients benefit from immunotherapy. In addition, cellular immunotherapies face important limitations imposed by the availability and quality of immune cells isolated from donors. Cell fate reprogramming is offering interesting alternatives to meet these challenges. Induced pluripotent stem cell (iPSC) technology not only enables studying immune cell specification but also serves as a platform for the differentiation of a myriad of clinically useful immune cells including T-cells, NK cells, or monocytes at scale. Moreover, the utilization of iPSCs allows introduction of genetic modifications and generation of T/NK cells with enhanced anti-tumor properties. Immune cells, such as macrophages and dendritic cells, can also be generated by direct cellular reprogramming employing lineage-specific master regulators bypassing the pluripotent stage. Thus, the cellular reprogramming toolbox is now providing the means to address the potential of patient-tailored immune cell types for cancer immunotherapy. In parallel, development of viral vectors for gene delivery has opened the door for in vivo reprogramming in regenerative medicine, an elegant strategy circumventing the current limitations of in vitro cell manipulation. An analogous paradigm has been recently developed in cancer immunotherapy by the generation of CAR T-cells in vivo. These new ideas on endogenous reprogramming, cross-fertilized from the fields of regenerative medicine and gene therapy, are opening exciting avenues for direct modulation of immune or tumor cells in situ, widening our strategies to remove cancer immunotherapy roadblocks. Here, we review current strategies for cancer immunotherapy, summarize technologies for generation of immune cells by cell fate reprogramming as well as highlight the future potential of inducing these unique cell identities in vivo, providing new and exciting tools for the fast-paced field of cancer immunotherapy.
Keywords: CAR-T; antigen presentation; cancer immunotherapy; cancer vaccine; cellular reprogramming; dendritic cell; transcription factor; tumor immunology.
Copyright © 2021 Zimmermannova, Caiado, Ferreira and Pereira.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical