

Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model
- PMID: 34369389
- PMCID: PMC8410092
- DOI: 10.1172/jci.insight.145991
Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model
Abstract
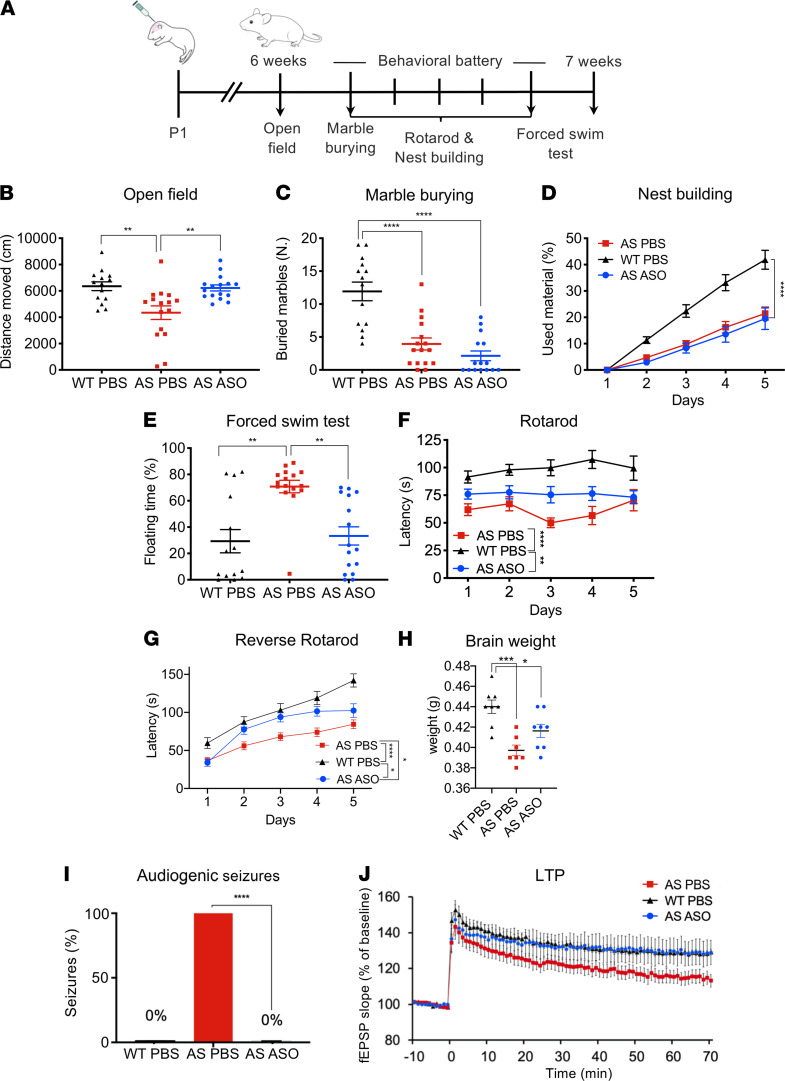
Angelman syndrome (AS) is a severe neurodevelopmental disorder for which only symptomatic treatment with limited benefits is available. AS is caused by mutations affecting the maternally inherited ubiquitin protein ligase E3A (UBE3A) gene. Previous studies showed that the silenced paternal Ube3a gene can be activated by targeting the antisense Ube3a-ATS transcript. We investigated antisense oligonucleotide-induced (ASO-induced) Ube3a-ATS degradation and its ability to induce UBE3A reinstatement and rescue of AS phenotypes in an established Ube3a mouse model. We found that a single intracerebroventricular injection of ASOs at postnatal day 1 (P1) or P21 in AS mice resulted in potent and specific UBE3A reinstatement in the brain, with levels up to 74% of WT levels in the cortex and a full rescue of sensitivity to audiogenic seizures. AS mice treated with ASO at P1 also showed rescue of established AS phenotypes, such as open field and forced swim test behaviors, and significant improvement on the reversed rotarod. Hippocampal plasticity of treated AS mice was comparable to WT but not significantly different from PBS-treated AS mice. No rescue was observed for the marble burying and nest building phenotypes. Our findings highlight the promise of ASO-mediated reactivation of UBE3A as a disease-modifying treatment for AS.
Keywords: Development; Mouse models; Neurodevelopment; Neurological disorders; Neuroscience.
Conflict of interest statement
Figures



References
-
- Williams CA, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet. 2006;140 A(5):413–418. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
