

Genome-wide sequencing as a first-tier screening test for short tandem repeat expansions
- PMID: 34372915
- PMCID: PMC8351082
- DOI: 10.1186/s13073-021-00932-9
Genome-wide sequencing as a first-tier screening test for short tandem repeat expansions
Erratum in
-
Correction to: Genome-wide sequencing as a first-tier screening test for short tandem repeat expansions.Genome Med. 2021 Sep 13;13(1):151. doi: 10.1186/s13073-021-00961-4. Genome Med. 2021. PMID: 34517885 Free PMC article. No abstract available.
Abstract
Background: Screening for short tandem repeat (STR) expansions in next-generation sequencing data can enable diagnosis, optimal clinical management/treatment, and accurate genetic counseling of patients with repeat expansion disorders. We aimed to develop an efficient computational workflow for reliable detection of STR expansions in next-generation sequencing data and demonstrate its clinical utility.
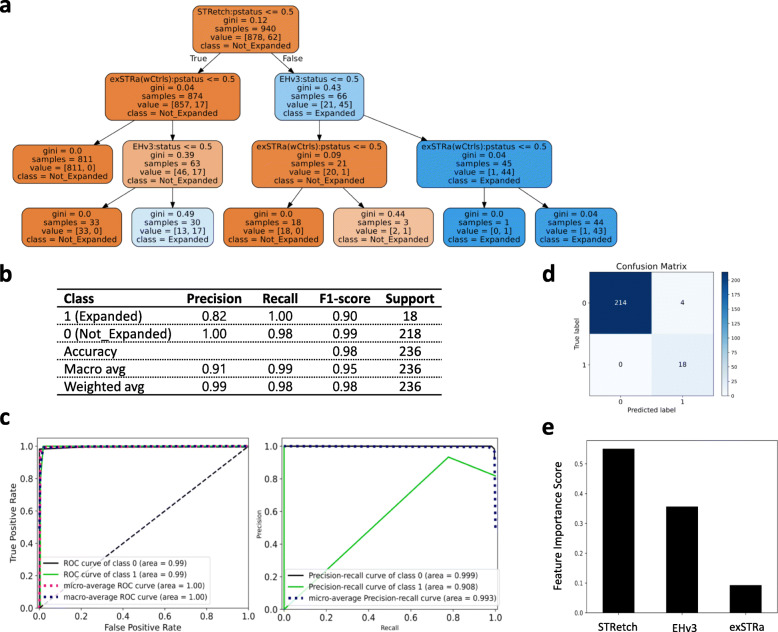
Methods: We characterized the performance of eight STR analysis methods (lobSTR, HipSTR, RepeatSeq, ExpansionHunter, TREDPARSE, GangSTR, STRetch, and exSTRa) on next-generation sequencing datasets of samples with known disease-causing full-mutation STR expansions and genomes simulated to harbor repeat expansions at selected loci and optimized their sensitivity. We then used a machine learning decision tree classifier to identify an optimal combination of methods for full-mutation detection. In Burrows-Wheeler Aligner (BWA)-aligned genomes, the ensemble approach of using ExpansionHunter, STRetch, and exSTRa performed the best (precision = 82%, recall = 100%, F1-score = 90%). We applied this pipeline to screen 301 families of children with suspected genetic disorders.
Results: We identified 10 individuals with full-mutations in the AR, ATXN1, ATXN8, DMPK, FXN, or HTT disease STR locus in the analyzed families. Additional candidates identified in our analysis include two probands with borderline ATXN2 expansions between the established repeat size range for reduced-penetrance and full-penetrance full-mutation and seven individuals with FMR1 CGG repeats in the intermediate/premutation repeat size range. In 67 probands with a prior negative clinical PCR test for the FMR1, FXN, or DMPK disease STR locus, or the spinocerebellar ataxia disease STR panel, our pipeline did not falsely identify aberrant expansion. We performed clinical PCR tests on seven (out of 10) full-mutation samples identified by our pipeline and confirmed the expansion status in all, showing absolute concordance between our bioinformatics and molecular findings.
Conclusions: We have successfully demonstrated the application of a well-optimized bioinformatics pipeline that promotes the utility of genome-wide sequencing as a first-tier screening test to detect expansions of known disease STRs. Interrogating clinical next-generation sequencing data for pathogenic STR expansions using our ensemble pipeline can improve diagnostic yield and enhance clinical outcomes for patients with repeat expansion disorders.
Keywords: Clinical bioinformatics; Machine learning; Next-generation sequencing; Repeat expansion; Short tandem repeats.
© 2021. The Author(s).
Conflict of interest statement
ED and MAE are employees of Illumina, Inc., a public company that develops and markets systems for genetic analysis. The remaining authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
