

NAD+ Redox Imbalance in the Heart Exacerbates Diabetic Cardiomyopathy
- PMID: 34374300
- PMCID: PMC8373812
- DOI: 10.1161/CIRCHEARTFAILURE.120.008170
NAD+ Redox Imbalance in the Heart Exacerbates Diabetic Cardiomyopathy
Abstract
Background: Diabetes is a risk factor for heart failure and promotes cardiac dysfunction. Diabetic tissues are associated with nicotinamide adenine dinucleotide (NAD+) redox imbalance; however, the hypothesis that NAD+ redox imbalance causes diabetic cardiomyopathy has not been tested. This investigation used mouse models with altered NAD+ redox balance to test this hypothesis.
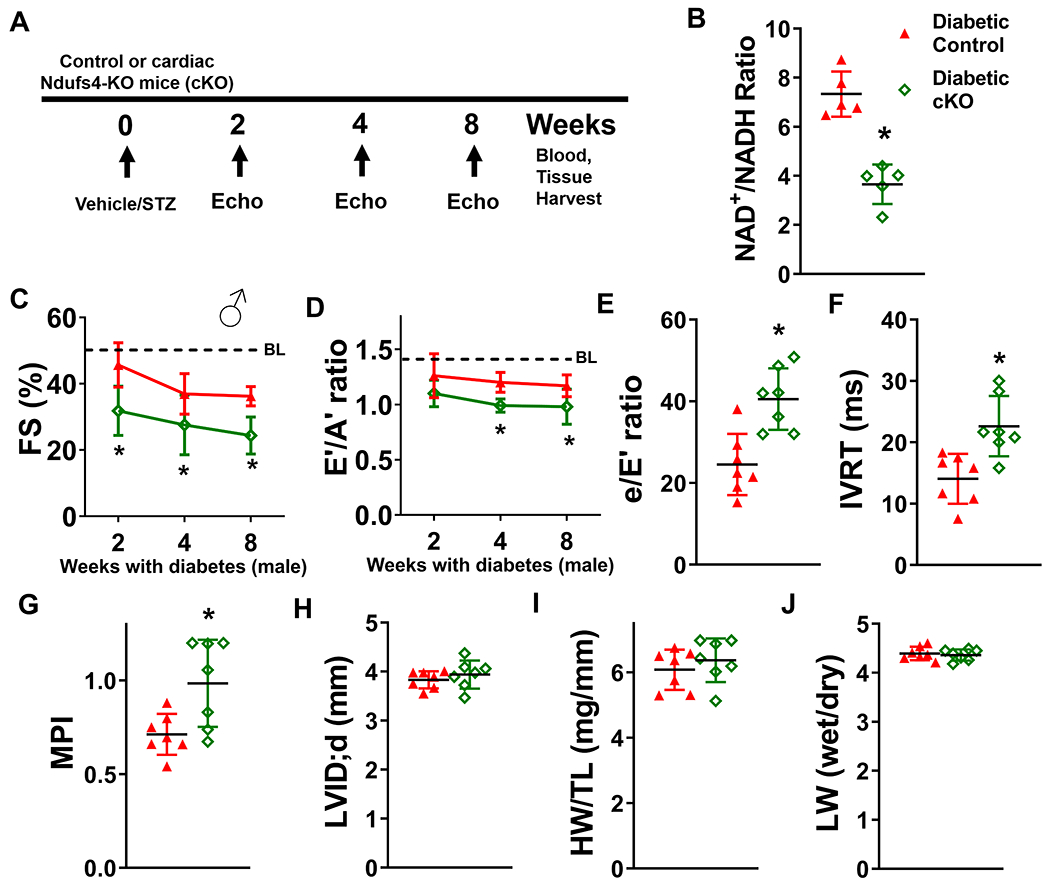
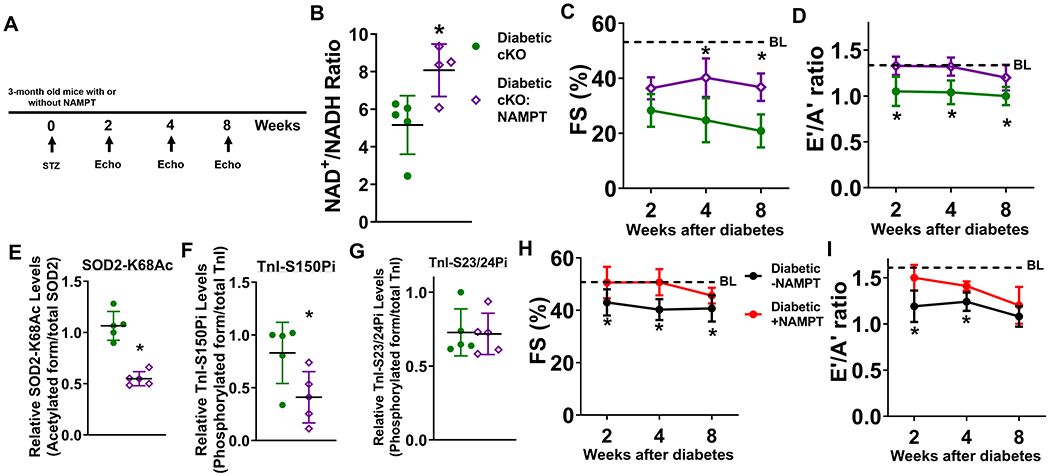
Methods: Diabetic stress was induced in mice by streptozotocin. Cardiac function was measured by echocardiography. Heart and plasma samples were collected for biochemical, histological, and molecular analyses. Two mouse models with altered NAD+ redox states (1, Ndufs4 [NADH:ubiquinone oxidoreductase subunit S4] knockout, cKO, and 2, NAMPT [nicotinamide phosphoribosyltranferase] transgenic mice, NMAPT) were used.
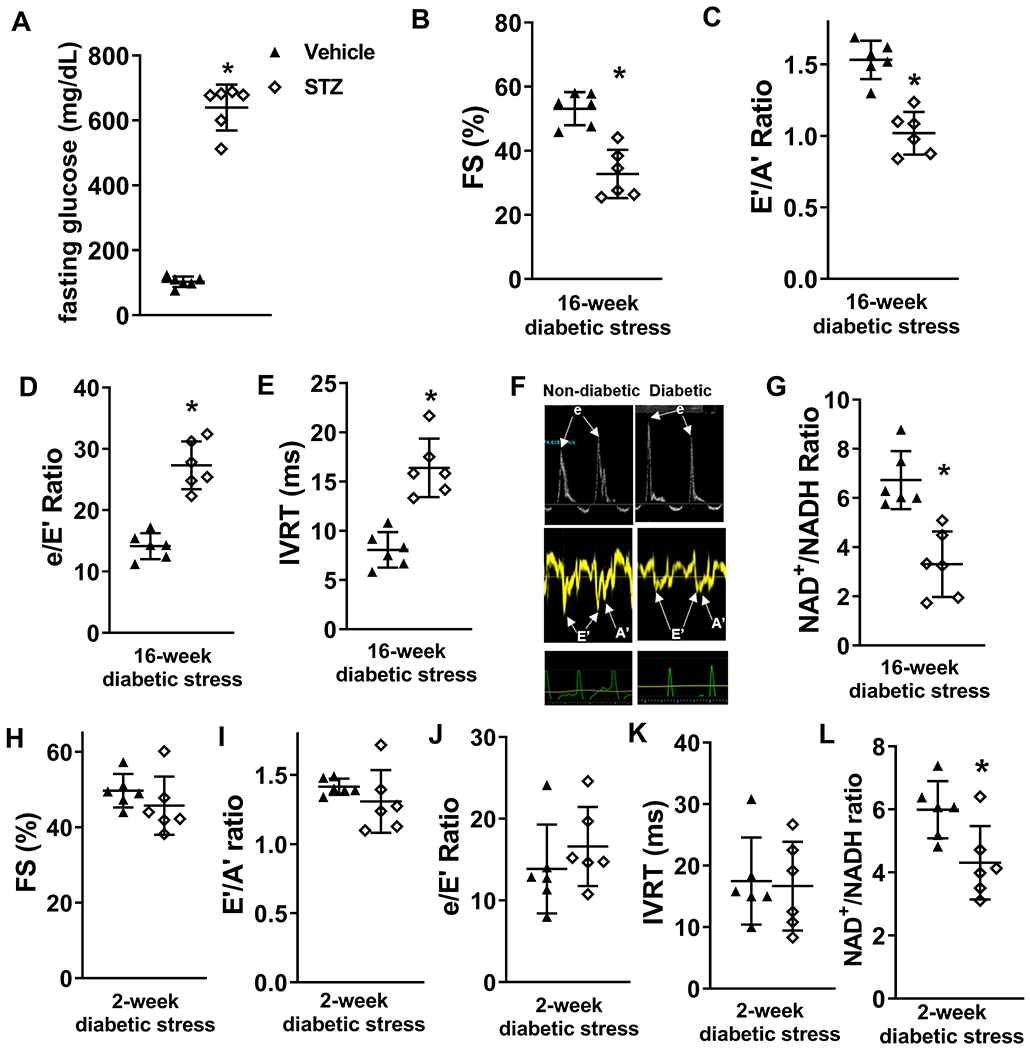
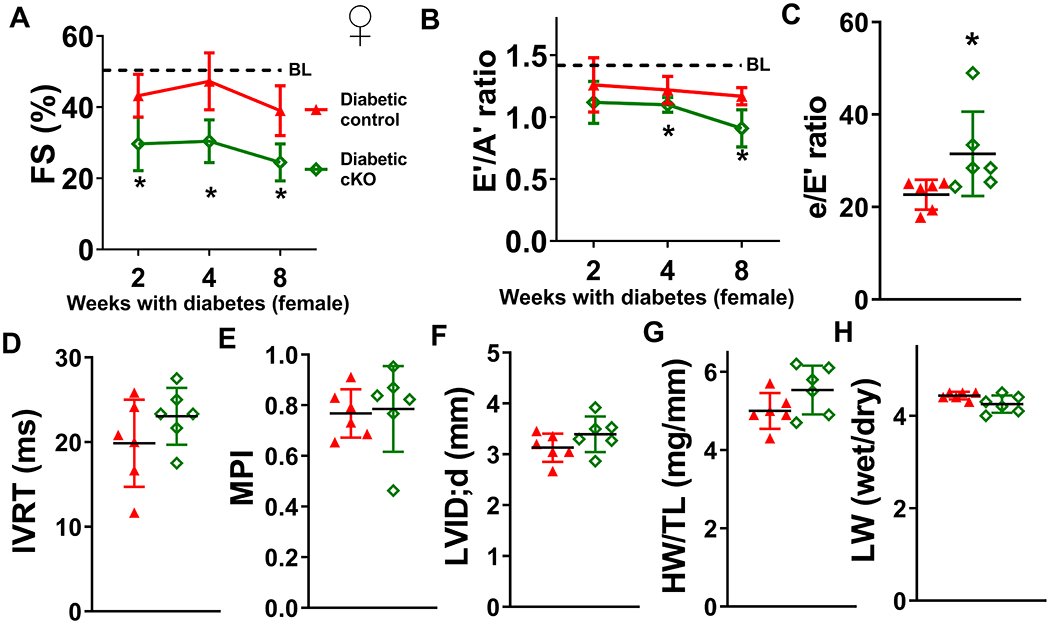
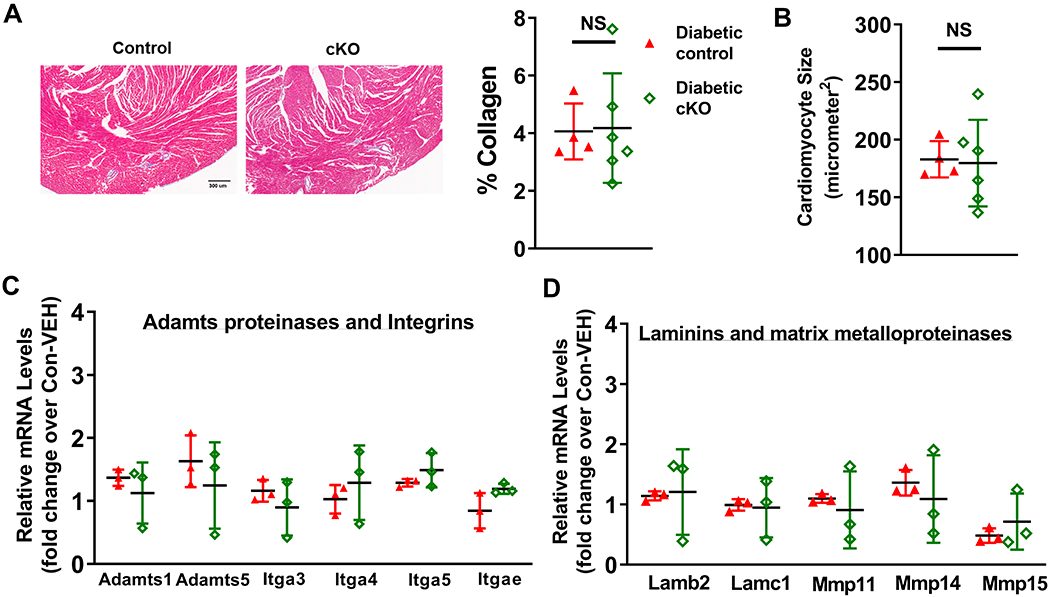
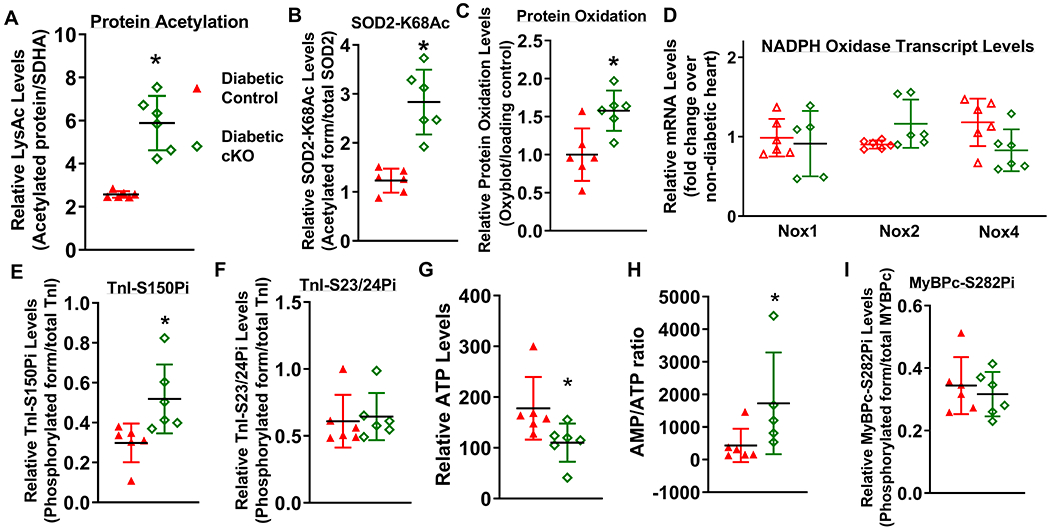
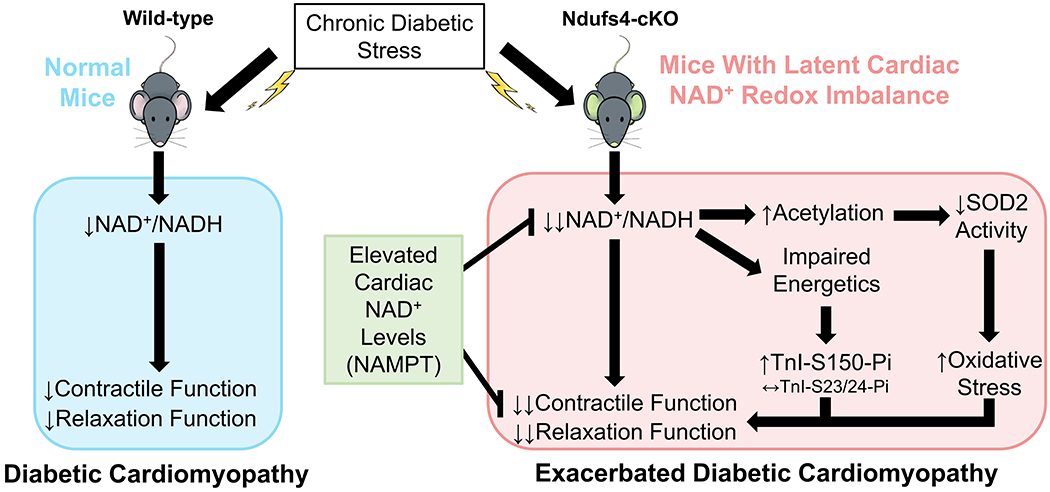
Results: Diabetic stress caused cardiac dysfunction and lowered NAD+/NADH ratio (oxidized/reduced ratio of nicotinamide adenine dinucleotide) in wild-type mice. Mice with lowered cardiac NAD+/NADH ratio without baseline dysfunction, cKO mice, were challenged with chronic diabetic stress. NAD+ redox imbalance in cKO hearts exacerbated systolic (fractional shortening: 27.6% versus 36.9% at 4 weeks, male cohort P<0.05), and diastolic dysfunction (early-to-late ratio of peak diastolic velocity: 0.99 versus 1.20, P<0.05) of diabetic mice in both sexes. Collagen levels and transcripts of fibrosis and extracellular matrix-dependent pathways did not show changes in diabetic cKO hearts, suggesting that the exacerbated cardiac dysfunction was due to cardiomyocyte dysfunction. NAD+ redox imbalance promoted superoxide dismutase 2 acetylation, protein oxidation, troponin I S150 phosphorylation, and impaired energetics in diabetic cKO hearts. Importantly, elevation of cardiac NAD+ levels by NAMPT normalized NAD+ redox balance, alleviated cardiac dysfunction (fractional shortening: 40.2% versus 24.8% in cKO:NAMPT versus cKO, P<0.05; early-to-late ratio of peak diastolic velocity: 1.32 versus 1.04, P<0.05), and reversed pathogenic mechanisms in diabetic mice.
Conclusions: Our results show that NAD+ redox imbalance to regulate acetylation and phosphorylation is a critical mediator of the progression of diabetic cardiomyopathy and suggest the therapeutic potential for diabetic cardiomyopathy by harnessing NAD+ metabolism.
Keywords: NAD+ redox imbalance; cardiomyopathy; diabetes; heart failure; risk factor.
Conflict of interest statement
Disclosures
No financial or non-financial competing interest is disclosed associated with the investigation in this manuscript by all authors.
Figures
References
-
- Choudhary C, Weinert BT, Nishida Y, Verdin E and Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nature reviews Molecular cell biology. 2014;15:536–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
