

Fused in sarcoma regulates DNA replication timing and kinetics
- PMID: 34375640
- PMCID: PMC8403768
- DOI: 10.1016/j.jbc.2021.101049
Fused in sarcoma regulates DNA replication timing and kinetics
Abstract
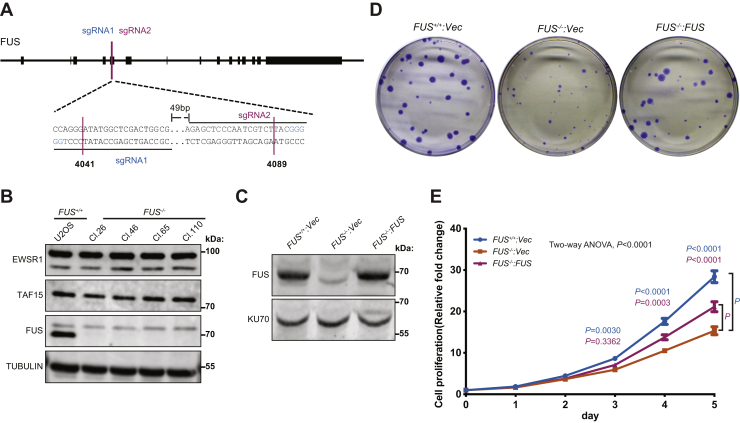
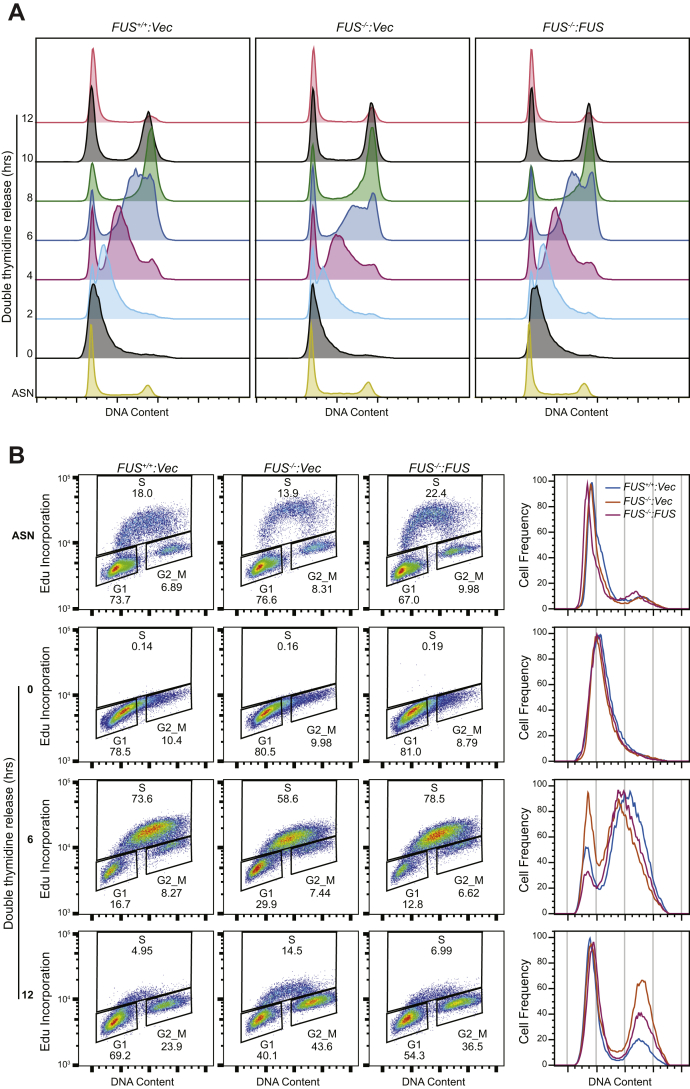
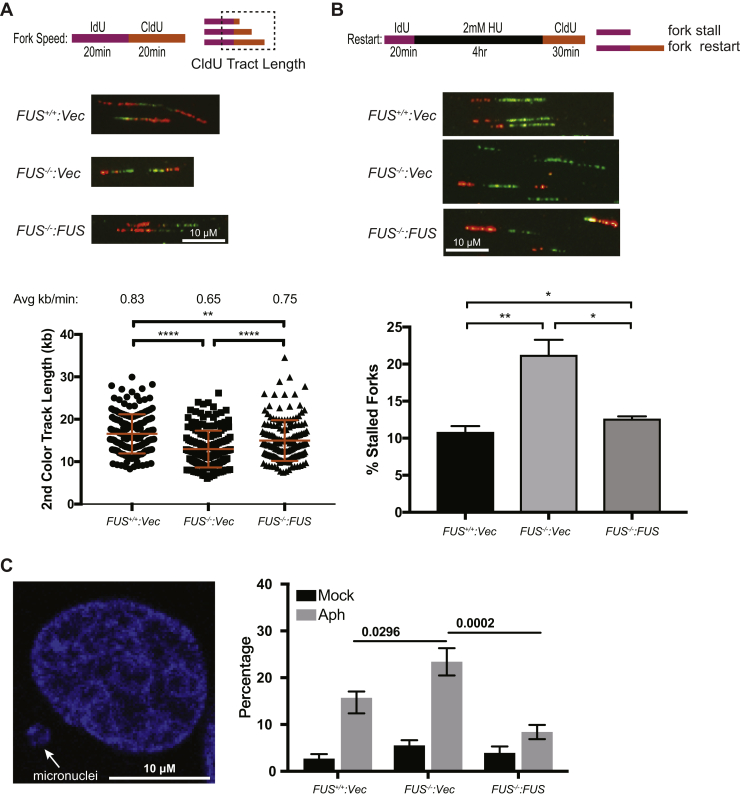
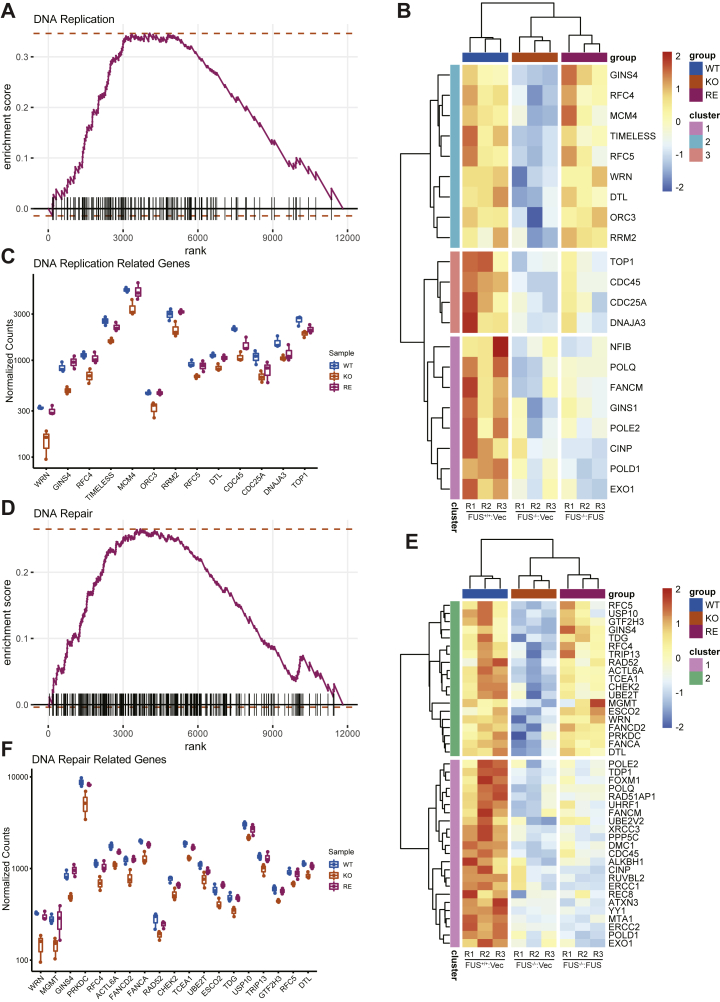
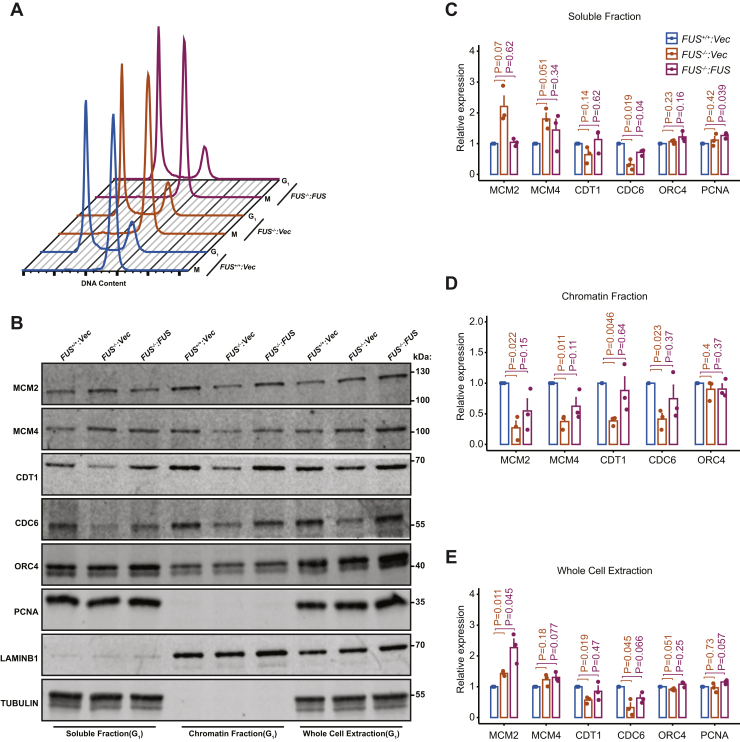
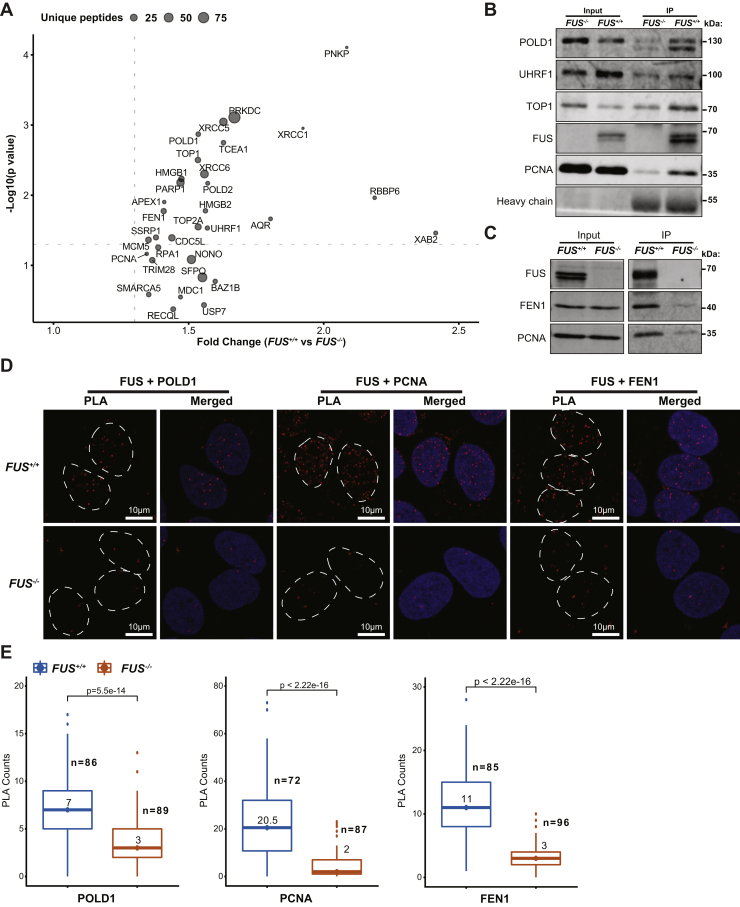
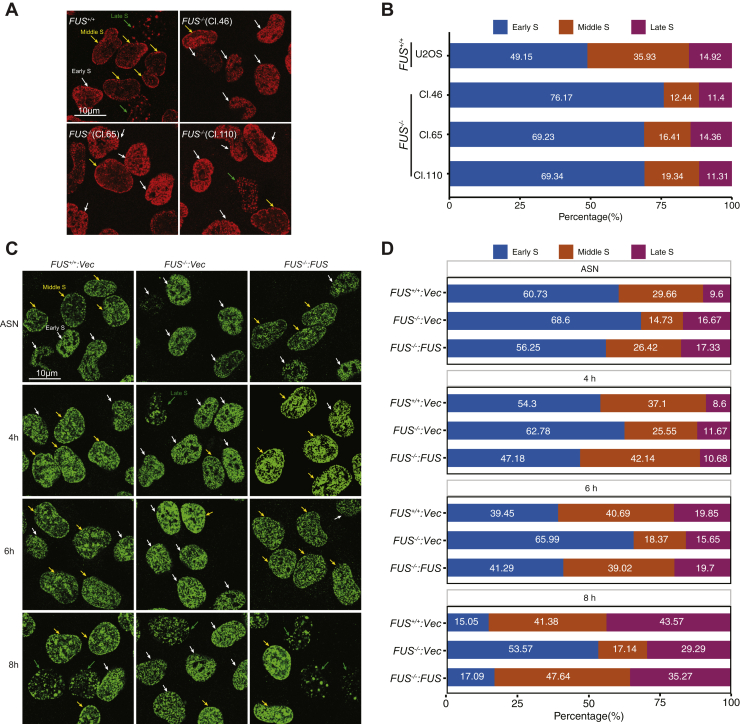
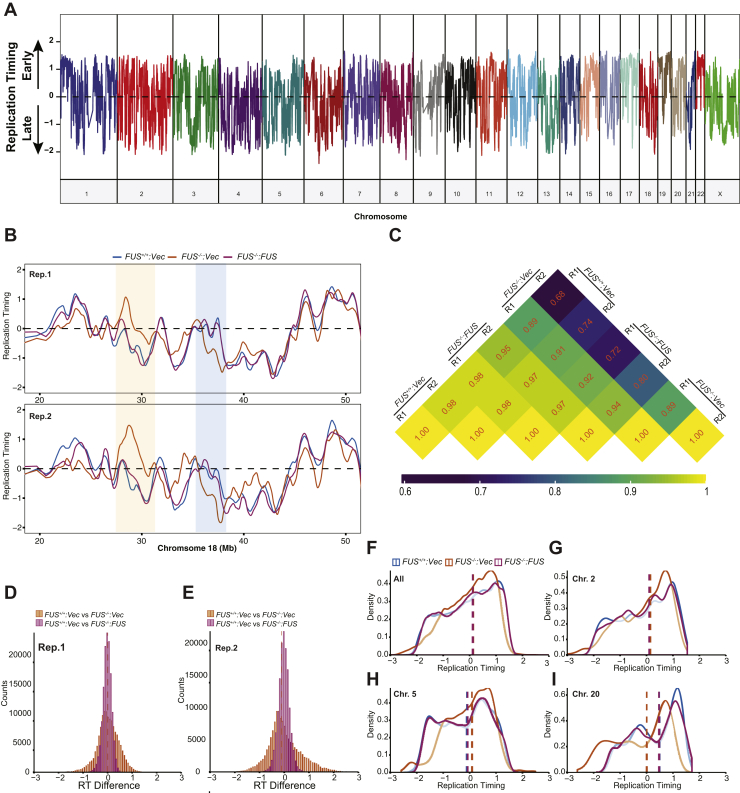
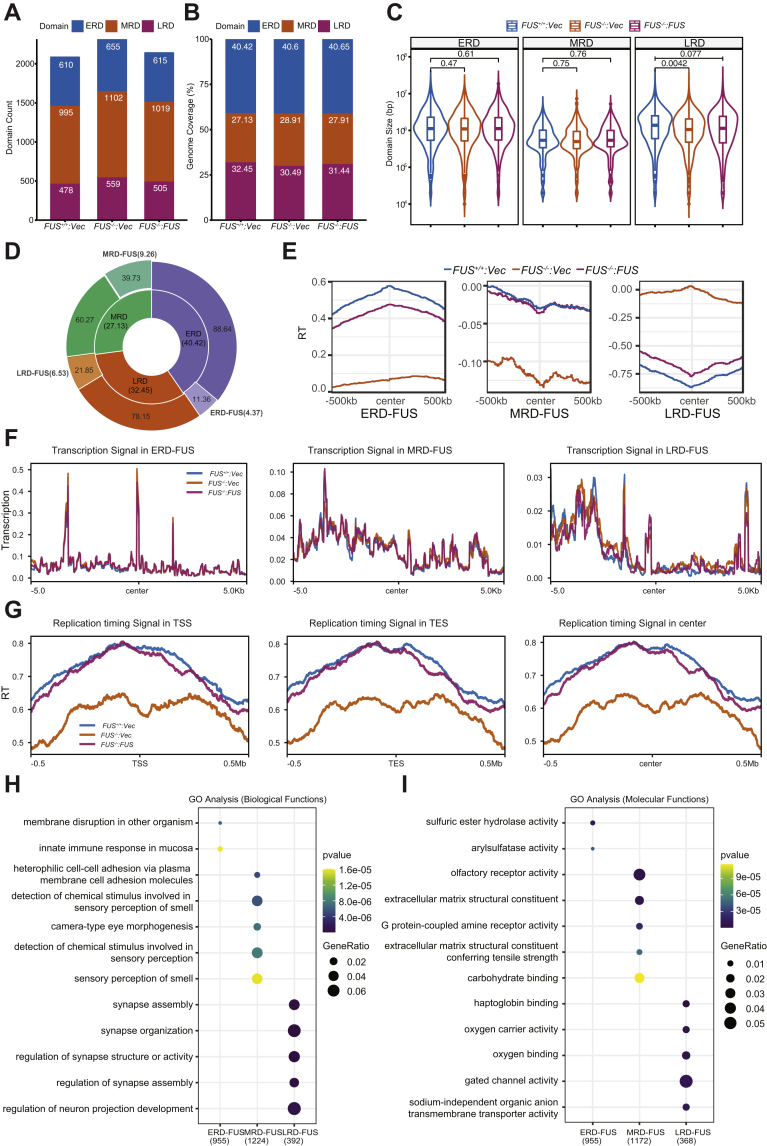
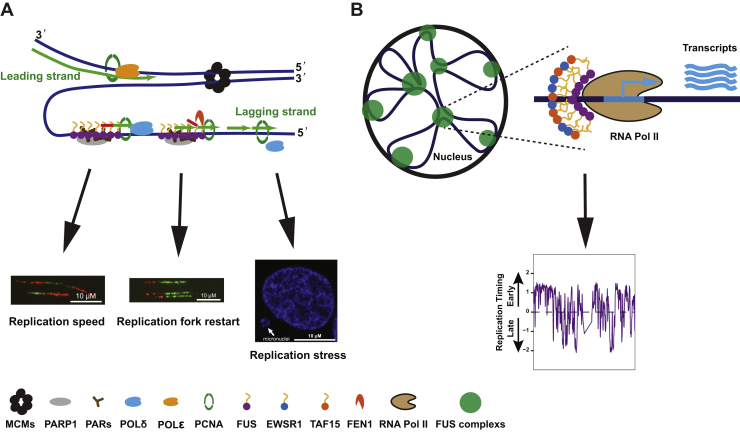
Fused in sarcoma (FUS) encodes an RNA-binding protein with diverse roles in transcriptional activation and RNA splicing. While oncogenic fusions of FUS and transcription factor DNA-binding domains are associated with soft tissue sarcomas, dominant mutations in FUS can cause amyotrophic lateral sclerosis. FUS has also been implicated in genome maintenance. However, the underlying mechanisms of its actions in genome stability are unknown. Here, we applied gene editing, functional reconstitution, and integrated proteomics and transcriptomics to illuminate roles for FUS in DNA replication and repair. Consistent with a supportive role in DNA double-strand break repair, FUS-deficient cells exhibited subtle alterations in the recruitment and retention of double-strand break-associated factors, including 53BP1 and BRCA1. FUS-/- cells also exhibited reduced proliferative potential that correlated with reduced speed of replication fork progression, diminished loading of prereplication complexes, enhanced micronucleus formation, and attenuated expression and splicing of S-phase-associated genes. Finally, FUS-deficient cells exhibited genome-wide alterations in DNA replication timing that were reversed upon re-expression of FUS complementary DNA. We also showed that FUS-dependent replication domains were enriched in transcriptionally active chromatin and that FUS was required for the timely replication of transcriptionally active DNA. These findings suggest that alterations in DNA replication kinetics and programming contribute to genome instability and functional defects in FUS-deficient cells.
Keywords: DNA repair; DNA replication; RNA binding protein; amyotrophic lateral sclerosis (ALS); fused in sarcoma (FUS); replication timing.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Rabbitts T.H., Forster A., Larson R., Nathan P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat. Genet. 1993;4:175–180. - PubMed
-
- Crozat A., Aman P., Mandahl N., Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature. 1993;363:640–644. - PubMed
-
- Kwiatkowski T.J., Jr., Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T., Valdmanis P., Rouleau G.A., Hosler B.A., Cortelli P., de Jong P.J. Mutations in the FUS/TLS on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
