

Rare variant contribution to human disease in 281,104 UK Biobank exomes
- PMID: 34375979
- PMCID: PMC8458098
- DOI: 10.1038/s41586-021-03855-y
Rare variant contribution to human disease in 281,104 UK Biobank exomes
Abstract
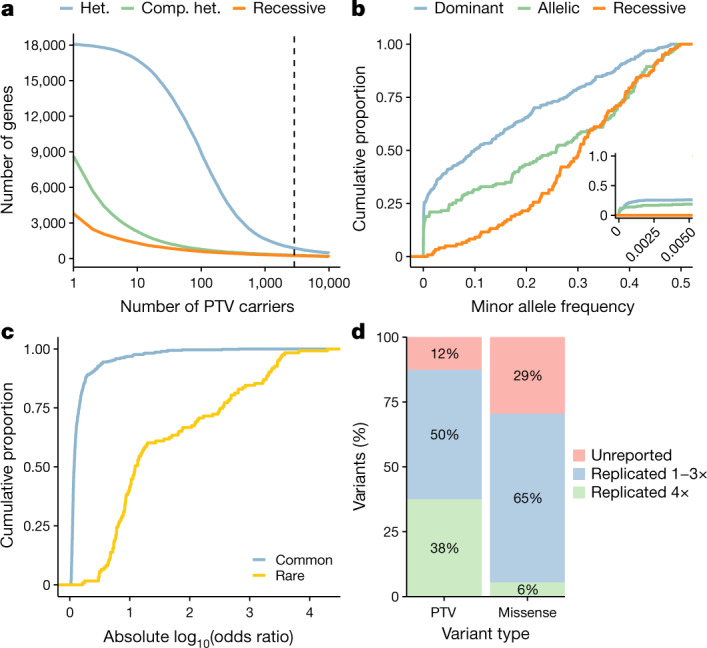
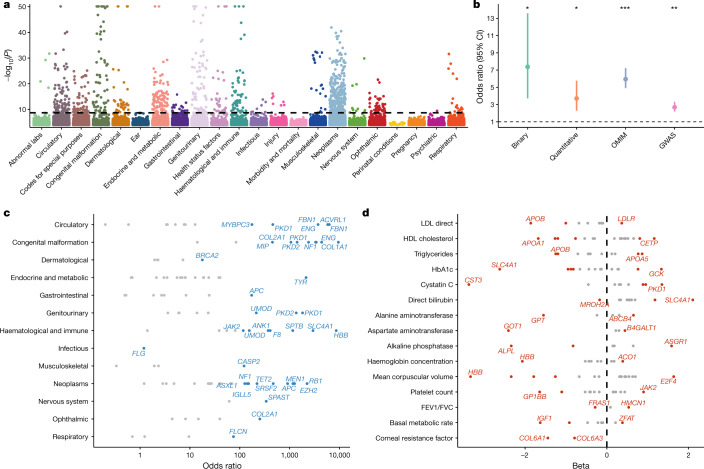
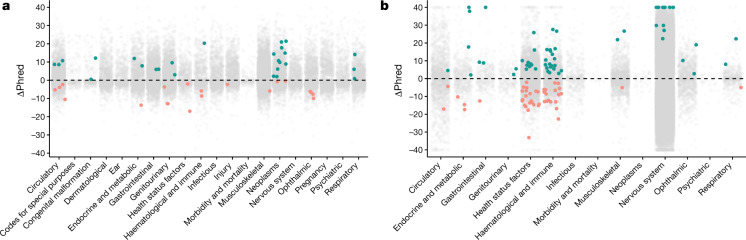
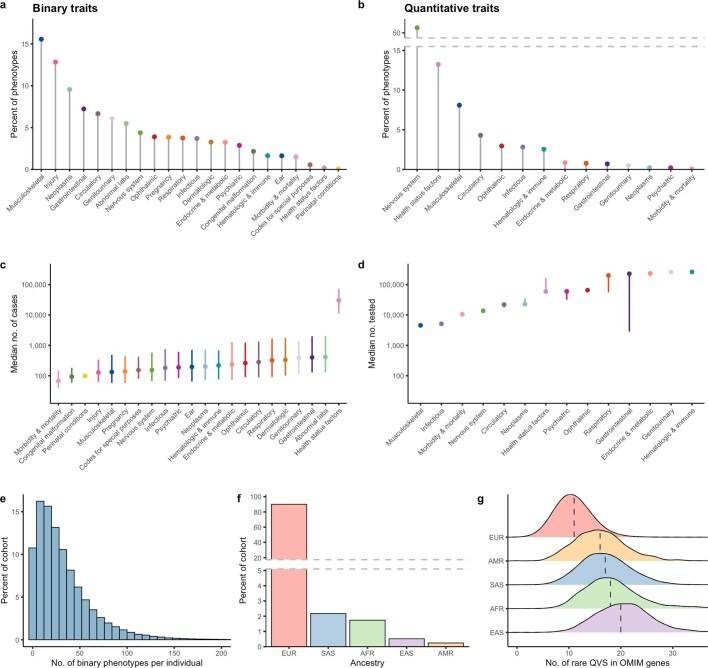
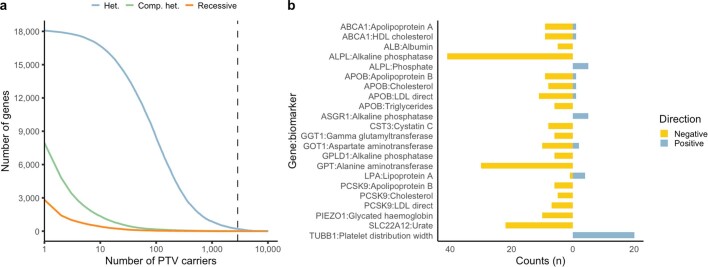
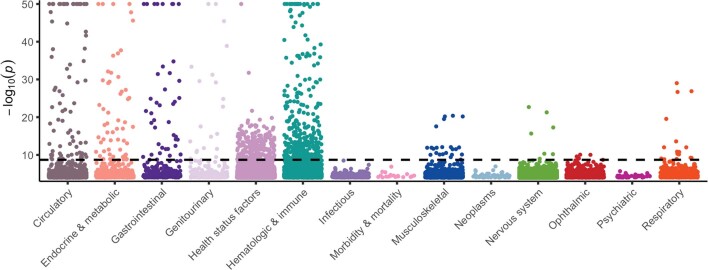
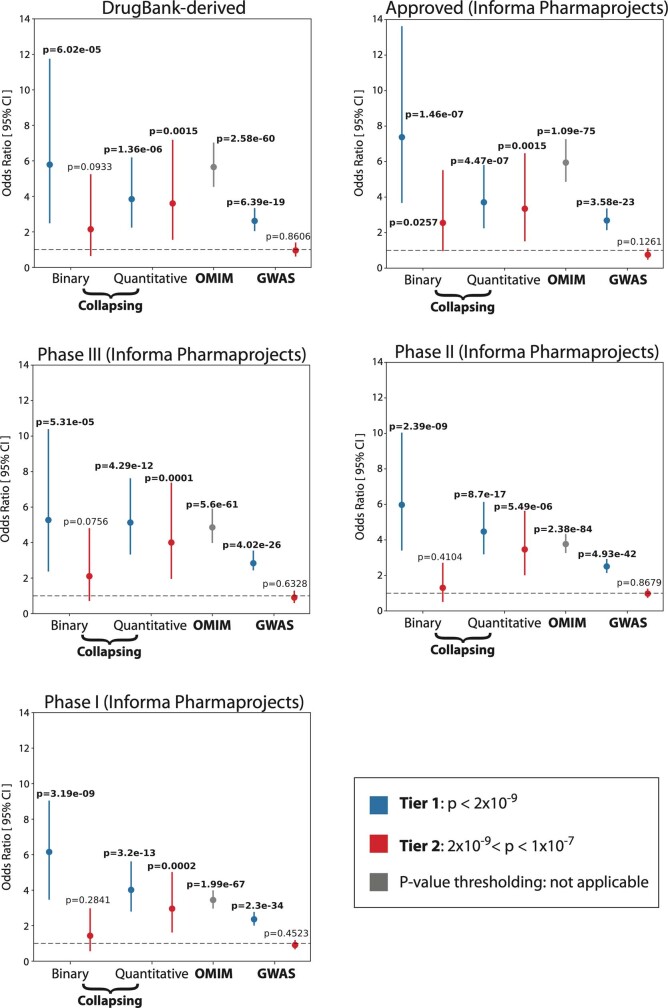
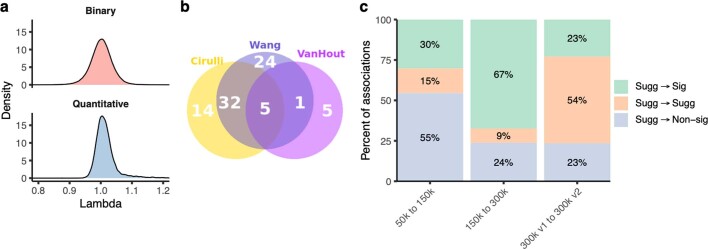
Genome-wide association studies have uncovered thousands of common variants associated with human disease, but the contribution of rare variants to common disease remains relatively unexplored. The UK Biobank contains detailed phenotypic data linked to medical records for approximately 500,000 participants, offering an unprecedented opportunity to evaluate the effect of rare variation on a broad collection of traits1,2. Here we study the relationships between rare protein-coding variants and 17,361 binary and 1,419 quantitative phenotypes using exome sequencing data from 269,171 UK Biobank participants of European ancestry. Gene-based collapsing analyses revealed 1,703 statistically significant gene-phenotype associations for binary traits, with a median odds ratio of 12.4. Furthermore, 83% of these associations were undetectable via single-variant association tests, emphasizing the power of gene-based collapsing analysis in the setting of high allelic heterogeneity. Gene-phenotype associations were also significantly enriched for loss-of-function-mediated traits and approved drug targets. Finally, we performed ancestry-specific and pan-ancestry collapsing analyses using exome sequencing data from 11,933 UK Biobank participants of African, East Asian or South Asian ancestry. Our results highlight a significant contribution of rare variants to common disease. Summary statistics are publicly available through an interactive portal ( http://azphewas.com/ ).
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Q.W., R.S.D., K.C., A.R.H., A.N., I.T., D.V., S.V.V.D., A.M., D.M., M.H., S.M., H.O., S.W., K.R.S., R.M., A.P., C.H. and S.P are current employees and/or stockholders of AstraZeneca.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
