

Triple-negative breast cancer: understanding Wnt signaling in drug resistance
- PMID: 34376211
- PMCID: PMC8353874
- DOI: 10.1186/s12935-021-02107-3
Triple-negative breast cancer: understanding Wnt signaling in drug resistance
Abstract
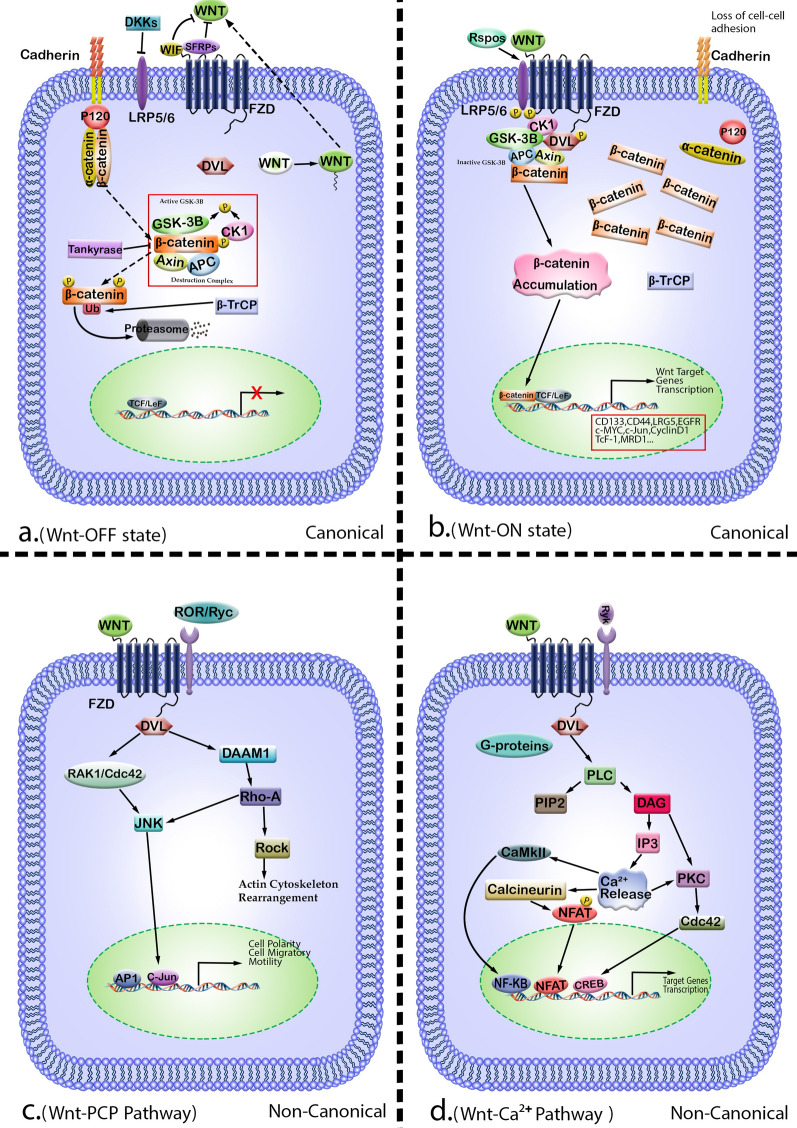
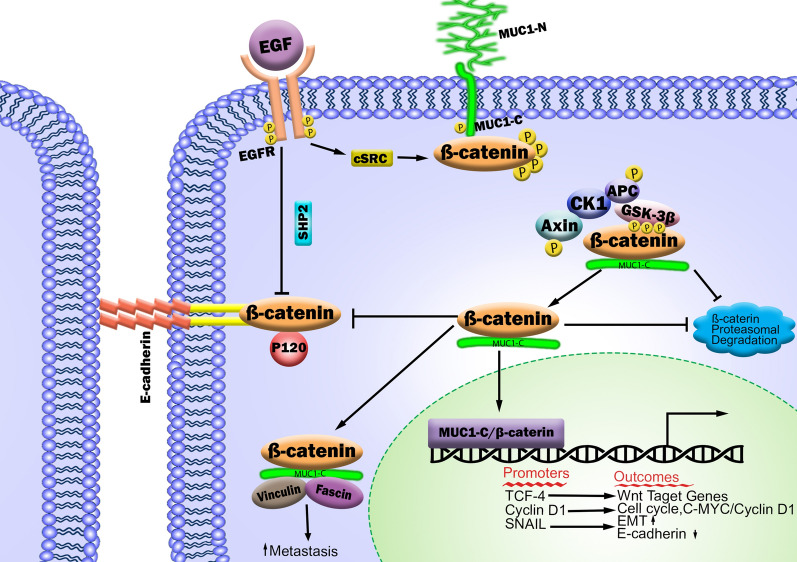
Triple-negative breast cancer (TNBC) is not as prevalent as hormone receptor or HER2-positive breast cancers and all receptor tests come back negative. More importantly, the heterogeneity and complexity of the TNBC on the molecular and clinical levels have limited the successful development of novel therapeutic strategies and led to intrinsic or developed resistance to chemotherapies and new therapeutic agents. Studies have demonstrated deregulation of Wnt/β-catenin signaling in tumorigenesis which plays decisive roles at the low survival rate of patients and facilitates resistance to currently existing therapies. This review summarizes mechanisms of Wnt/β-catenin signaling for resistance development in TNBC, the complex interaction between Wnt/β-catenin signaling, and the transactivated receptor tyrosine kinase (RTK) signaling pathways, lymphocytic infiltration, epithelial-mesenchymal transition (EMT), and induction of metastasis. Such associations and how these pathways interact in the development and progression of cancer have led to the careful analysis and development of new and effective combination therapies without generating significant toxicity and resistance.
Keywords: Combination therapy; Drug resistance; Triple negative breast cancers (TNBCs); Tumorigenesis; Wnt/β-catenin.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
The natural compound Jatrophone interferes with Wnt/β-catenin signaling and inhibits proliferation and EMT in human triple-negative breast cancer.PLoS One. 2017 Dec 27;12(12):e0189864. doi: 10.1371/journal.pone.0189864. eCollection 2017. PLoS One. 2017. PMID: 29281678 Free PMC article.
-
Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states.Mol Oncol. 2018 Apr;12(4):423-440. doi: 10.1002/1878-0261.12167. Epub 2018 Feb 21. Mol Oncol. 2018. PMID: 29316250 Free PMC article.
-
Low Dose of Paclitaxel Combined with XAV939 Attenuates Metastasis, Angiogenesis and Growth in Breast Cancer by Suppressing Wnt Signaling.Cells. 2019 Aug 14;8(8):892. doi: 10.3390/cells8080892. Cells. 2019. PMID: 31416135 Free PMC article.
-
Countering Triple Negative Breast Cancer via Impeding Wnt/β-Catenin Signaling, a Phytotherapeutic Approach.Plants (Basel). 2022 Aug 24;11(17):2191. doi: 10.3390/plants11172191. Plants (Basel). 2022. PMID: 36079579 Free PMC article. Review.
-
Potential therapeutic targets of triple-negative breast cancer based on its intrinsic subtype.Oncotarget. 2017 Aug 16;8(42):73329-73344. doi: 10.18632/oncotarget.20274. eCollection 2017 Sep 22. Oncotarget. 2017. PMID: 29069872 Free PMC article. Review.
Cited by
-
Network medicine based approach for identifying the type 2 diabetes, osteoarthritis and triple negative breast cancer interactome: Finding the hub of hub genes.Heliyon. 2024 Aug 22;10(17):e36650. doi: 10.1016/j.heliyon.2024.e36650. eCollection 2024 Sep 15. Heliyon. 2024. PMID: 39281650 Free PMC article.
-
Interference KRT17 reverses doxorubicin resistance in triple-negative breast cancer cells by Wnt/β-catenin signaling pathway.Genes Genomics. 2023 Oct;45(10):1329-1338. doi: 10.1007/s13258-023-01437-y. Epub 2023 Aug 27. Genes Genomics. 2023. PMID: 37634232 Free PMC article.
-
Investigating the role of exosomal long non-coding RNAs in drug resistance within female reproductive system cancers.Front Cell Dev Biol. 2025 Jan 24;13:1485422. doi: 10.3389/fcell.2025.1485422. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 39925739 Free PMC article. Review.
-
The Role of PAX7 in Breast Cancer Prognosis and Its Mechanistic Involvement in the Wnt/β-Catenin Pathway.J Cell Mol Med. 2025 May;29(10):e70602. doi: 10.1111/jcmm.70602. J Cell Mol Med. 2025. PMID: 40370330 Free PMC article.
-
Harnessing adrenergic blockade in stress-promoted TNBC in vitro and solid tumor in vivo: disrupting HIF-1α and GSK-3β/β-catenin driven resistance to doxorubicin.Front Pharmacol. 2024 Jun 19;15:1362675. doi: 10.3389/fphar.2024.1362675. eCollection 2024. Front Pharmacol. 2024. PMID: 38962320 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
