

DFT and MD simulation investigation of favipiravir as an emerging antiviral option against viral protease (3CLpro) of SARS-CoV-2
- PMID: 34376872
- PMCID: PMC8342190
- DOI: 10.1016/j.molstruc.2021.131253
DFT and MD simulation investigation of favipiravir as an emerging antiviral option against viral protease (3CLpro) of SARS-CoV-2
Abstract
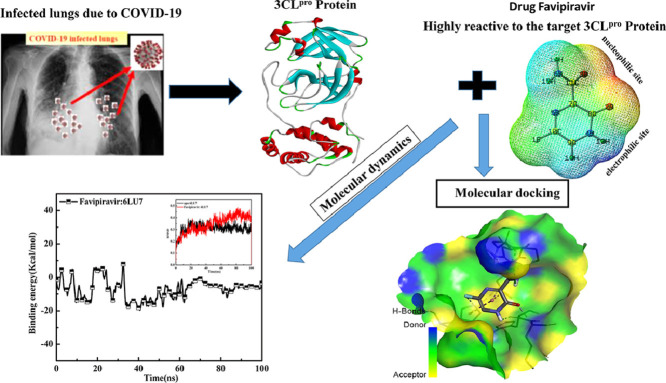
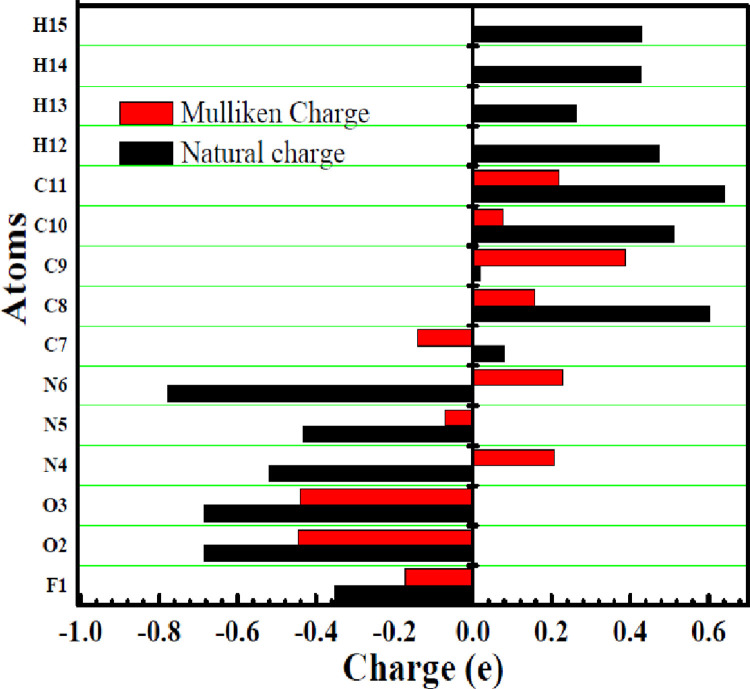
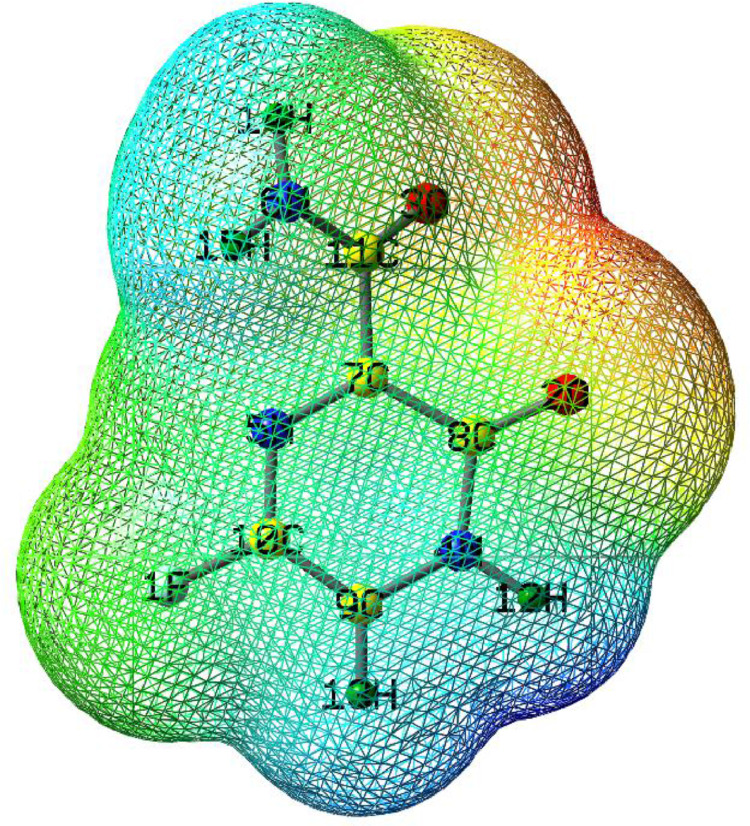
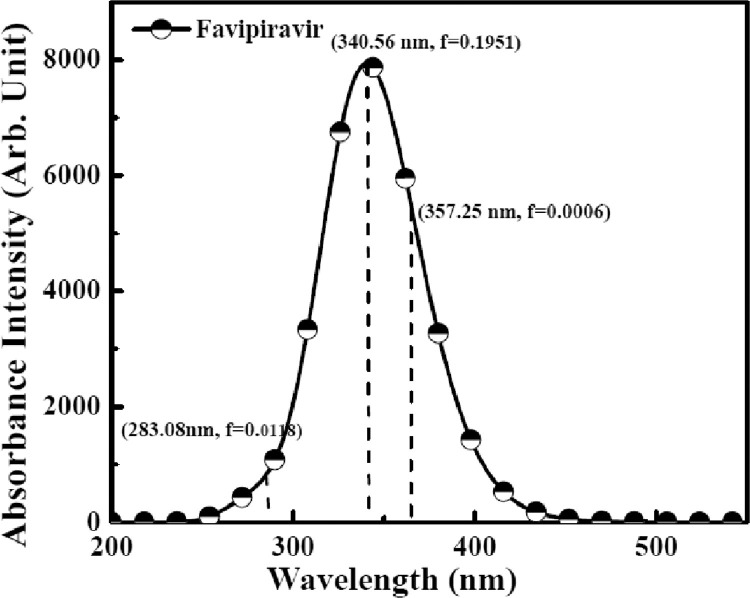
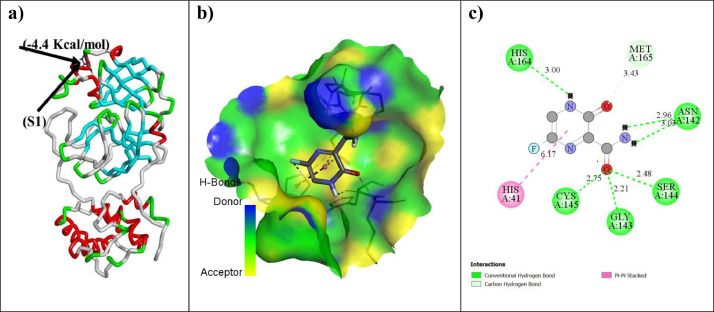
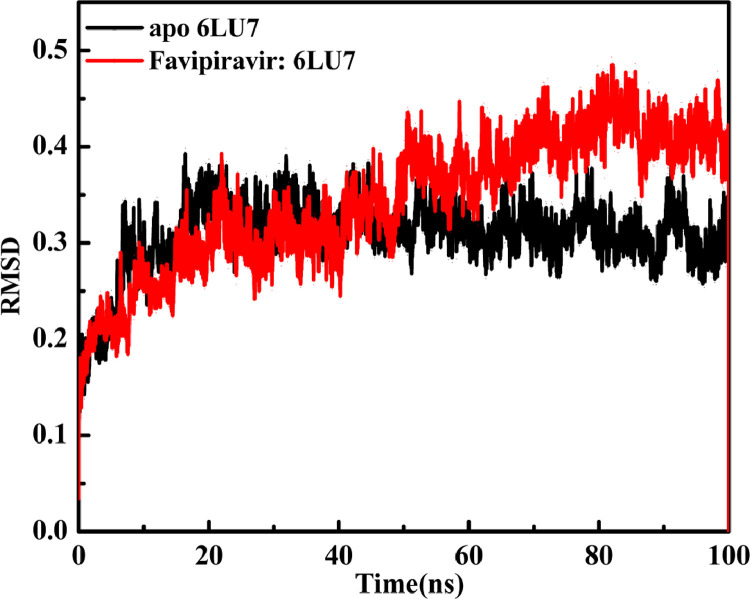
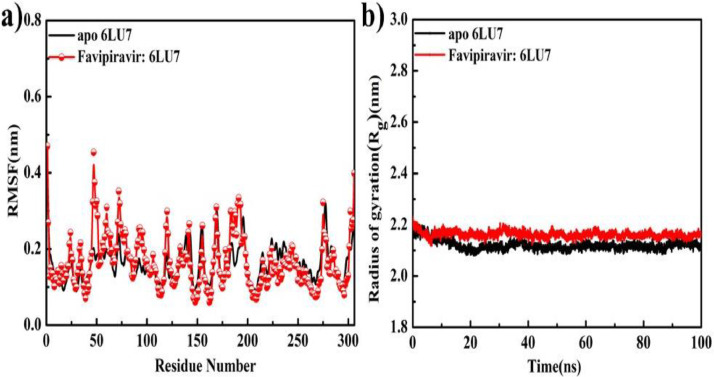
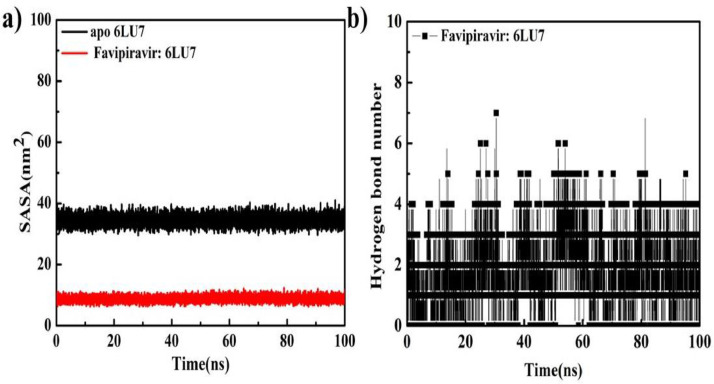
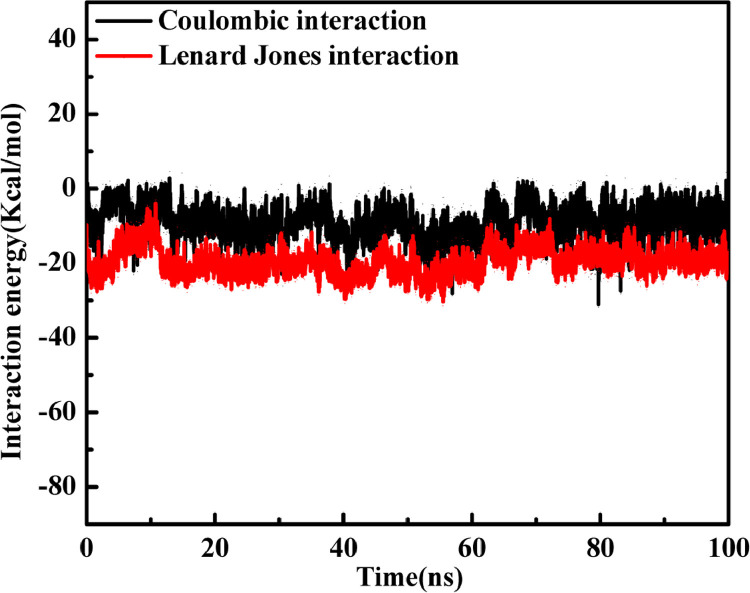
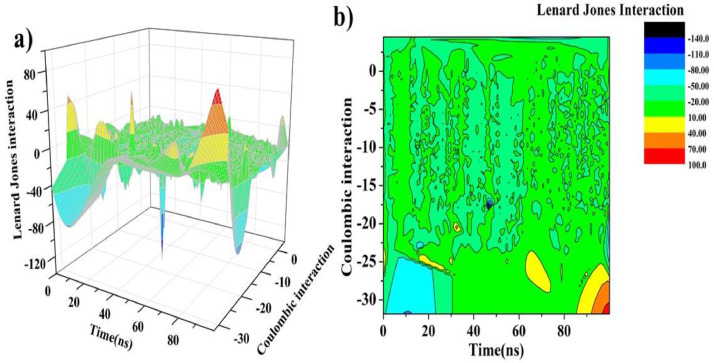
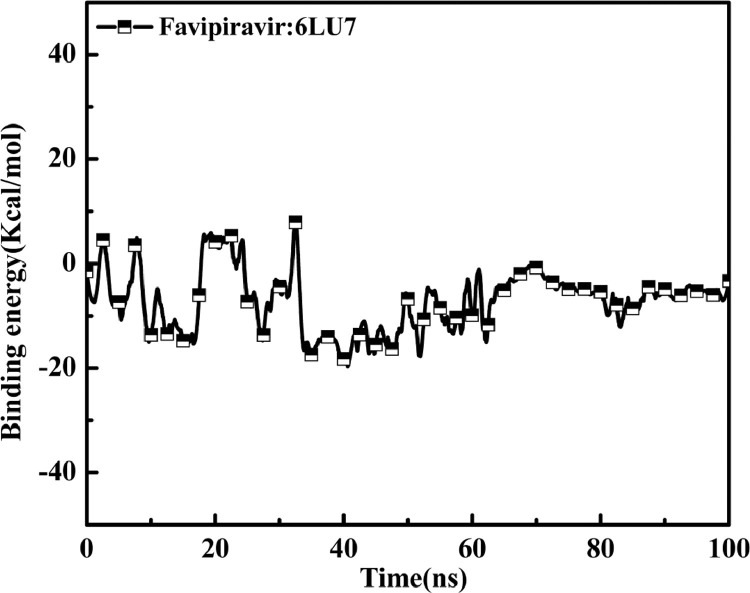
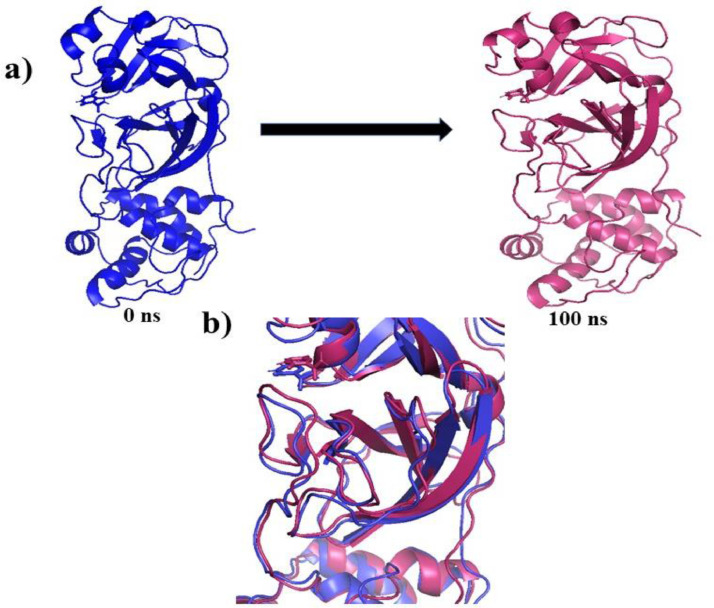
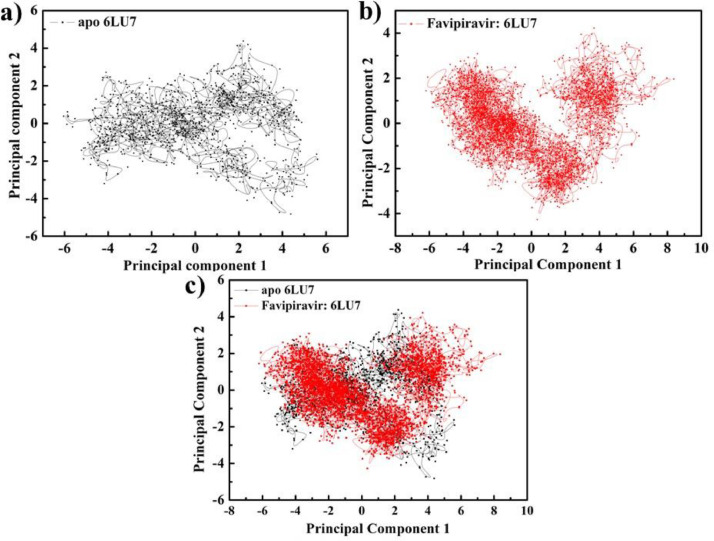
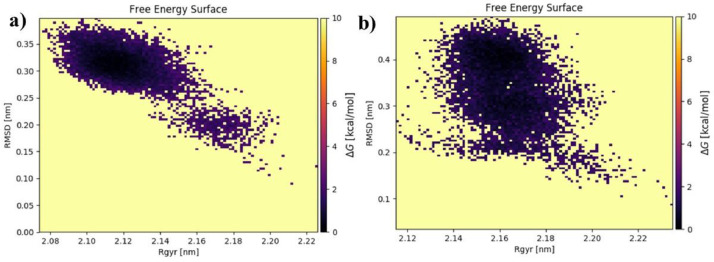
As per date, around 20 million COVID-19 cases reported from across the globe due to a tiny 125 nm sized virus: SARS-CoV-2 which has created a pandemic and left an unforgettable impact on our world. Besides vaccine, medical community is in a race to identify an effective drug, which can fight against this disease effectively. Favipiravir (F) has recently attracted too much attention as an effective repurposed drug against COVID-19. In the present study, the pertinency of F has been tested as an antiviral option against viral protease (3CLpro) of SARS-CoV-2 with the help of density functional theory (DFT) and MD Simulation. Different electronic properties of F such as atomic charges, molecular electrostatic properties (MEP), chemical reactivity and absorption analysis have been studied by DFT. In order to understand the interaction and stability of inhibitor F against viral protease, molecular docking and MD simulation have been performed. Various output like interaction energies, number of intermolecular hydrogen bonding, binding energy etc. have established the elucidate role of F for the management of CoV-2 virus for which there is no approved therapies till now. Our findings highlighted the need to further evaluate F as a potential antiviral against SARS-CoV-2.
Keywords: Density functional theory; Electronic properties; Favipiravir; Molecular docking; SARS-CoV-2.
© 2021 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Gorbalenya A.E., Baker S.C., Ralph S.B., de Groot R.J., Drosten C., Gulyaeva A.A., Haagmans B.L., Leontovich A.M., Neuman B.W., Penzar D., Poon L.L.M., Samborskiy D.V., Sidorov I., Sola I., Ziebuhr J. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020 doi: 10.1038/s41564-020-0695-z. - DOI - PMC - PubMed
-
- Aytür Y.K., Köseoğlu B.F., Taşkıran Ö.Ö., Ordu-Gökkaya N.K., Delialioğlu S.Ü., Tur B.S, Sarıkaya S., Şirzai H., Tiftik T.T., Alemdaroğlu E., Ayhan F.F. Pulmonary rehabilitation principles in SARS-COV-2 infection (COVID-19): a guideline for the acute and subacute rehabilitation. Turk. J. Phys. Med. Rehabil. 2020;66(2):104. - PMC - PubMed
-
- Skariyachan S., Gopal D., Chakrabarti S., Kempanna P., Uttarkar A., Muddebihalkar A.G., Niranjan V. Structural and molecular basis of the interaction mechanism of selected drugs towards multiple targets of SARS-CoV-2 by molecular docking and dynamic simulation studies-deciphering the scope of repurposed drugs. Comput. Biol. Med. 2020;1(126) - PMC - PubMed
-
- Wang B., Guo H., Ling L., Ji J., Niu J., Gu Y. The chronic adverse effect of chloroquine on kidney in rats through an autophagy dependent and independent pathways. Nephron. 2020;144(1):53–64. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
