

Biallelic truncating variants in ATP9A cause a novel neurodevelopmental disorder involving postnatal microcephaly and failure to thrive
- PMID: 34379057
- PMCID: PMC9252857
- DOI: 10.1136/jmedgenet-2021-107843
Biallelic truncating variants in ATP9A cause a novel neurodevelopmental disorder involving postnatal microcephaly and failure to thrive
Abstract
Background: Genes implicated in the Golgi and endosomal trafficking machinery are crucial for brain development, and mutations in them are particularly associated with postnatal microcephaly (POM).
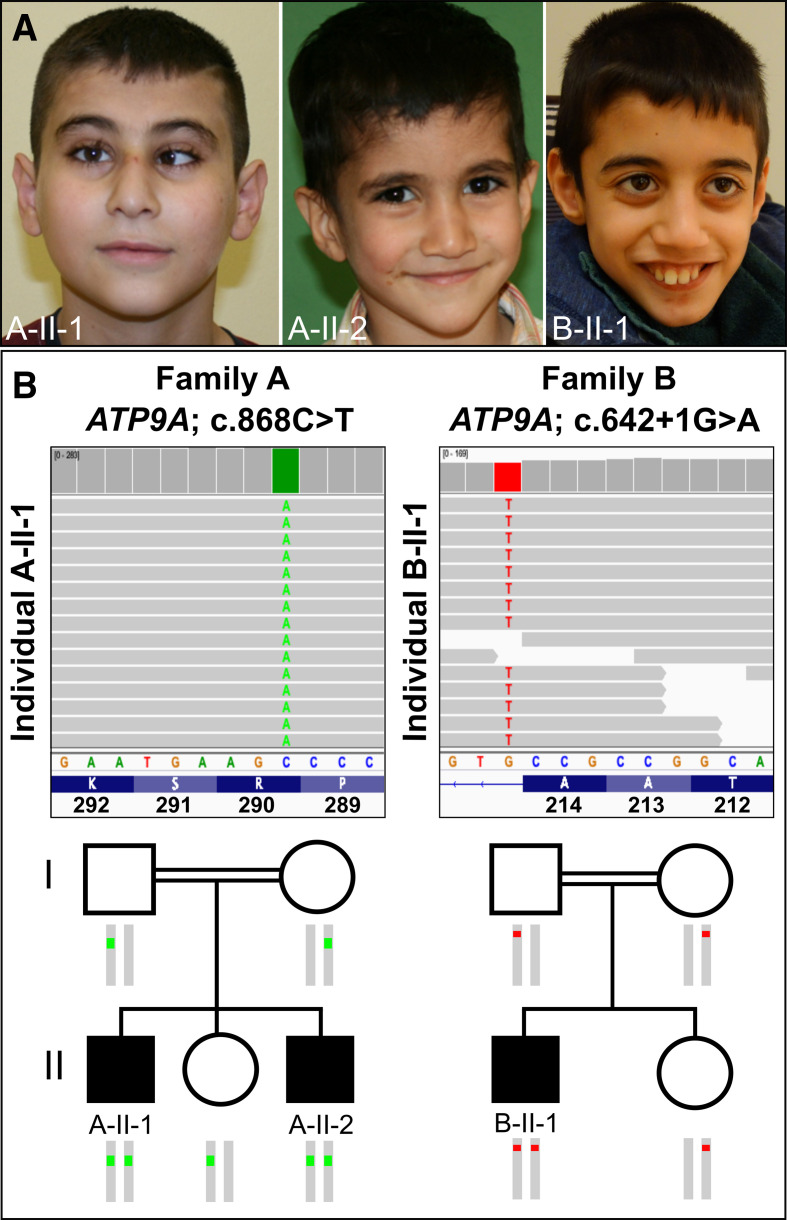
Methods: Exome sequencing was performed in three affected individuals from two unrelated consanguineous families presenting with delayed neurodevelopment, intellectual disability of variable degree, POM and failure to thrive. Patient-derived fibroblasts were tested for functional effects of the variants.
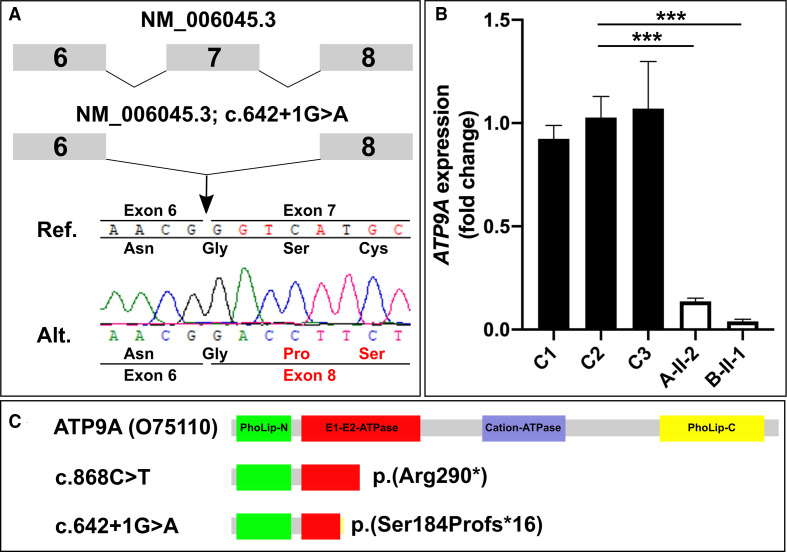
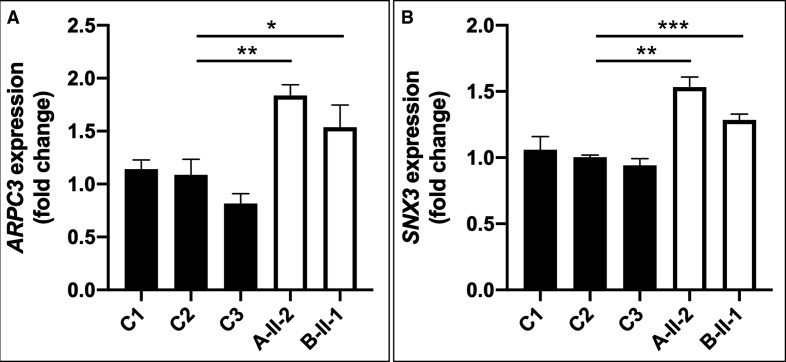
Results: We detected homozygous truncating variants in ATP9A. While the variant in family A is predicted to result in an early premature termination codon, the variant in family B affects a canonical splice site. Both variants lead to a substantial reduction of ATP9A mRNA expression. It has been shown previously that ATP9A localises to early and recycling endosomes, whereas its depletion leads to altered gene expression of components from this compartment. Consistent with previous findings, we also observed overexpression of ARPC3 and SNX3, genes strongly interacting with ATP9A.
Conclusion: In aggregate, our findings show that pathogenic variants in ATP9A cause a novel autosomal recessive neurodevelopmental disorder with POM. While the physiological function of endogenous ATP9A is still largely elusive, our results underline a crucial role of this gene in endosomal transport in brain tissue.
Keywords: DNA; RNA; and neonatal diseases and abnormalities; congenital; genetics; hereditary; medical; sequence analysis.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Liu P, Meng L, Normand EA, Xia F, Song X, Ghazi A, Rosenfeld J, Magoulas PL, Braxton A, Ward P, Dai H, Yuan B, Bi W, Xiao R, Wang X, Chiang T, Vetrini F, He W, Cheng H, Dong J, Gijavanekar C, Benke PJ, Bernstein JA, Eble T, Eroglu Y, Erwin D, Escobar L, Gibson JB, Gripp K, Kleppe S, Koenig MK, Lewis AM, Natowicz M, Mancias P, Minor L, Scaglia F, Schaaf CP, Streff H, Vernon H, Uhles CL, Zackai EH, Wu N, Sutton VR, Beaudet AL, Muzny D, Gibbs RA, Posey JE, Lalani S, Shaw C, Eng CM, Lupski JR, Yang Y. Reanalysis of clinical exome sequencing data. N Engl J Med 2019;380:2478–80. 10.1056/NEJMc1812033 - DOI - PMC - PubMed
-
- Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F, Keerthikumar S, Oortveld MAW, Kleefstra T, Kramer JM, Webber C, Huynen MA, Schenck A. Systematic phenomics analysis Deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet 2016;98:149–64. 10.1016/j.ajhg.2015.11.024 - DOI - PMC - PubMed
-
- Boonsawat P, Joset P, Steindl K, Oneda B, Gogoll L, Azzarello-Burri S, Sheth F, Datar C, Verma IC, Puri RD, Zollino M, Bachmann-Gagescu R, Niedrist D, Papik M, Figueiro-Silva J, Masood R, Zweier M, Kraemer D, Lincoln S, Rodan L, Passemard S, Drunat S, Verloes A, Horn AHC, Sticht H, Steinfeld R, Plecko B, Latal B, Jenni O, Asadollahi R, Rauch A, Undiagnosed Diseases Network (UDN) . Elucidation of the phenotypic spectrum and genetic landscape in primary and secondary microcephaly. Genet Med 2019;21:2043–58. 10.1038/s41436-019-0464-7 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous