

FACT is recruited to the +1 nucleosome of transcribed genes and spreads in a Chd1-dependent manner
- PMID: 34380014
- PMCID: PMC9149603
- DOI: 10.1016/j.molcel.2021.07.010
FACT is recruited to the +1 nucleosome of transcribed genes and spreads in a Chd1-dependent manner
Abstract
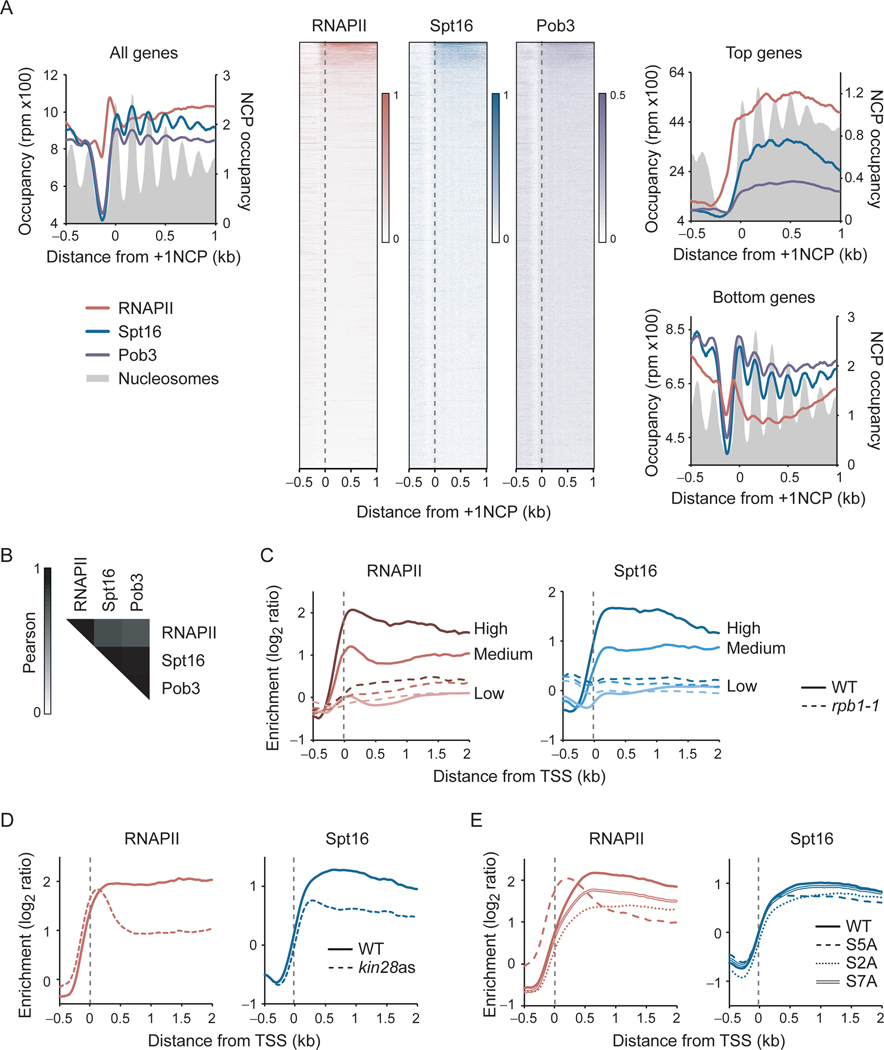
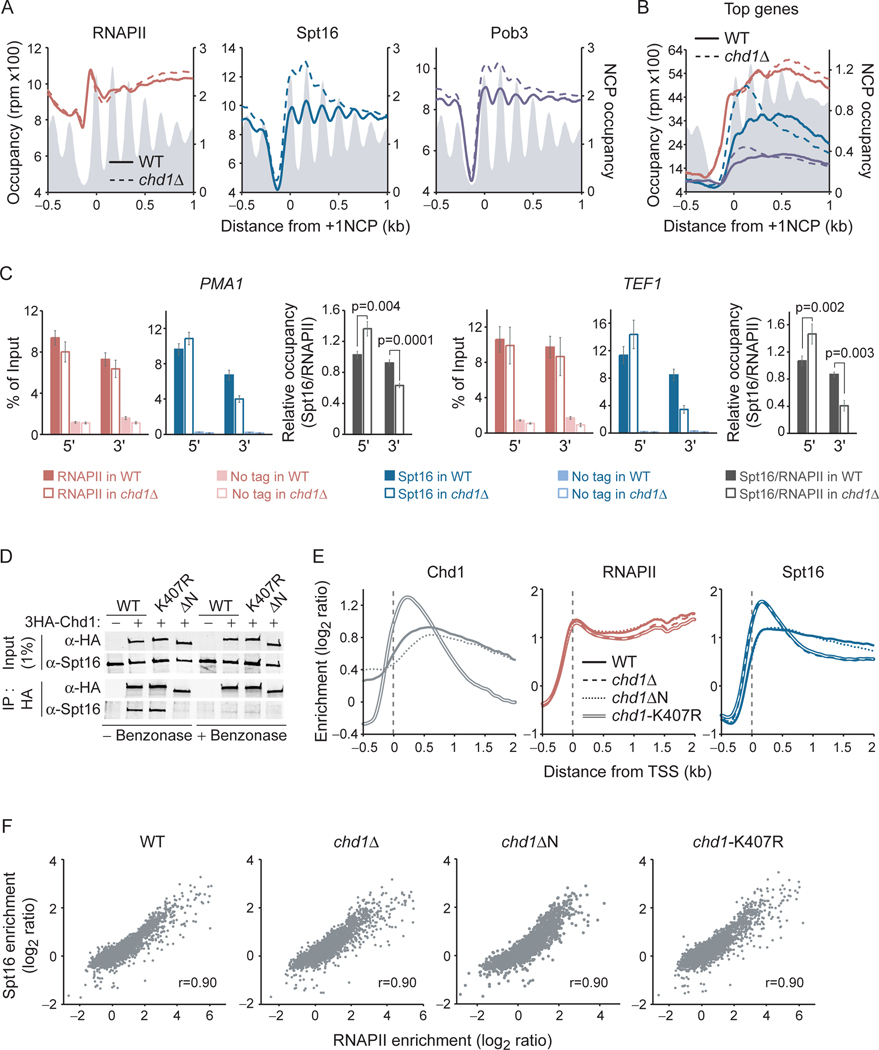
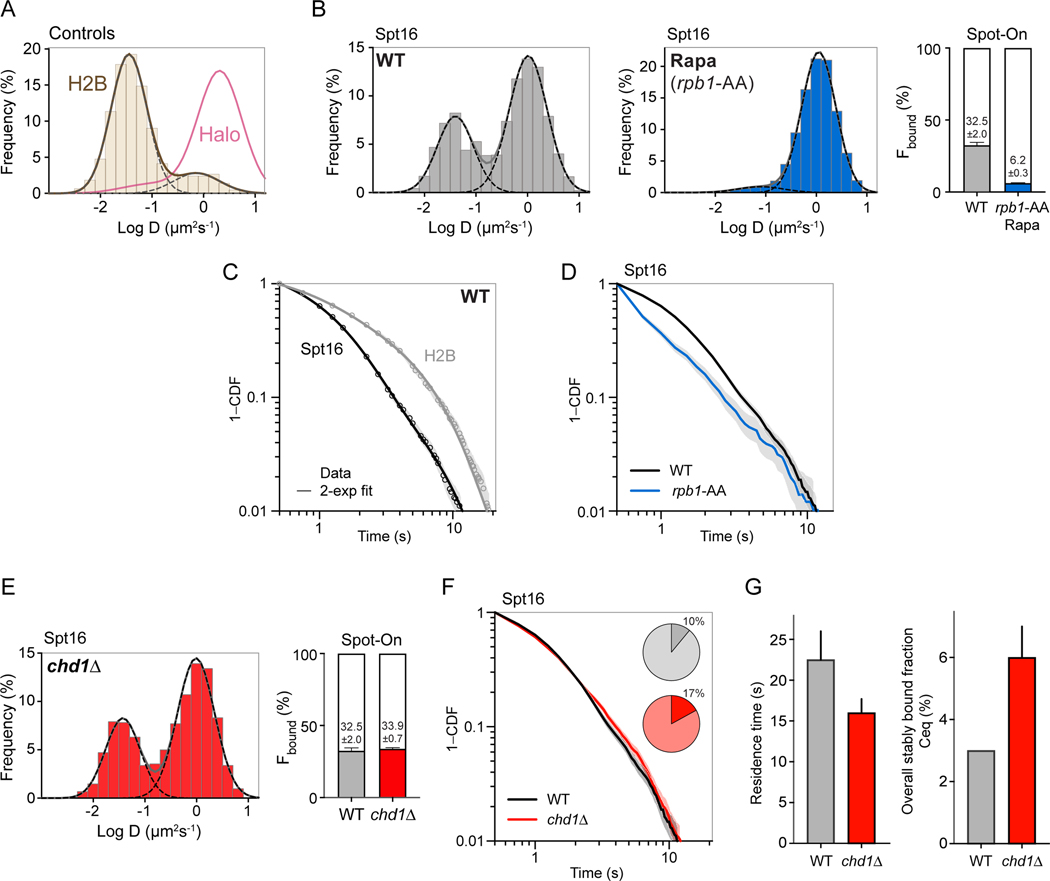
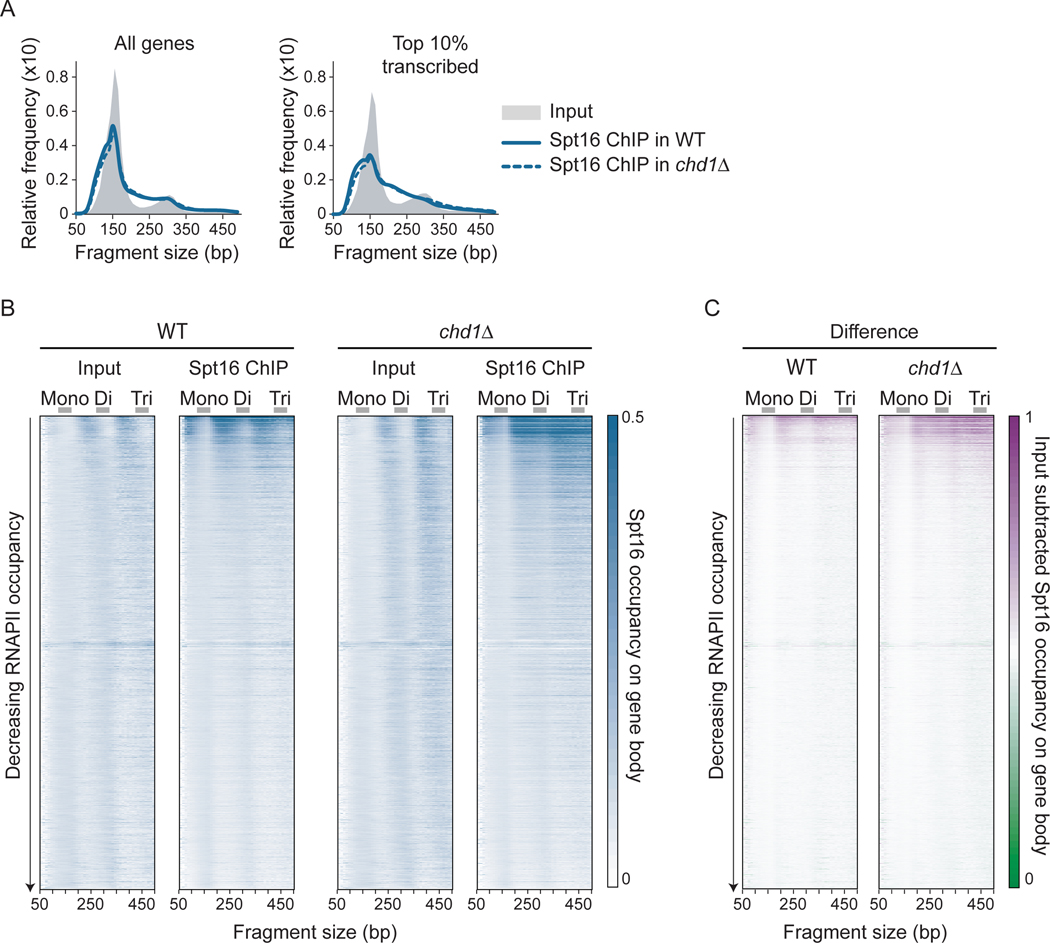
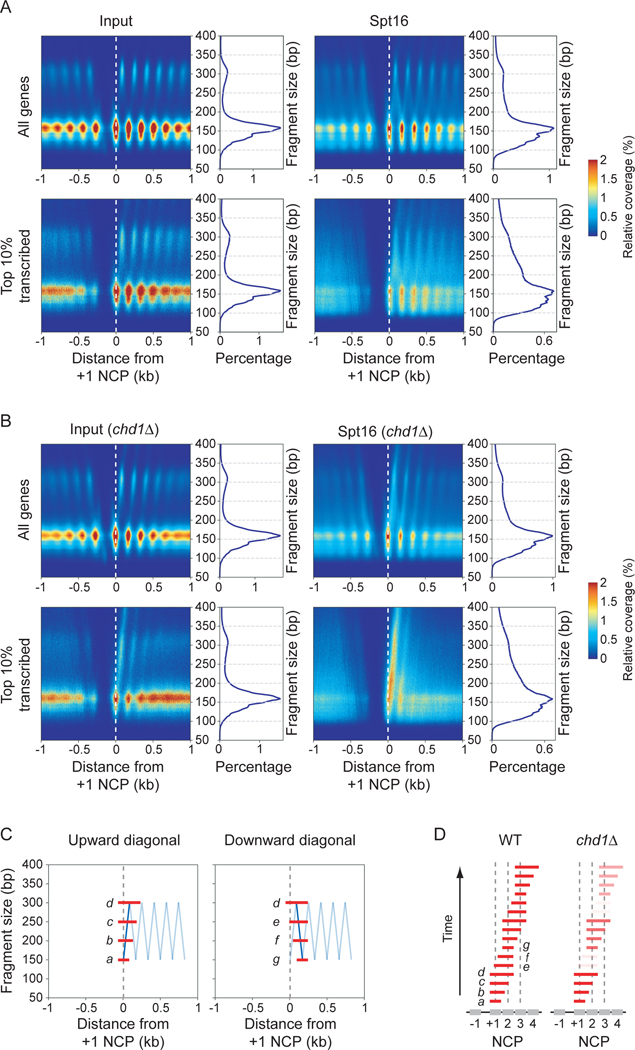
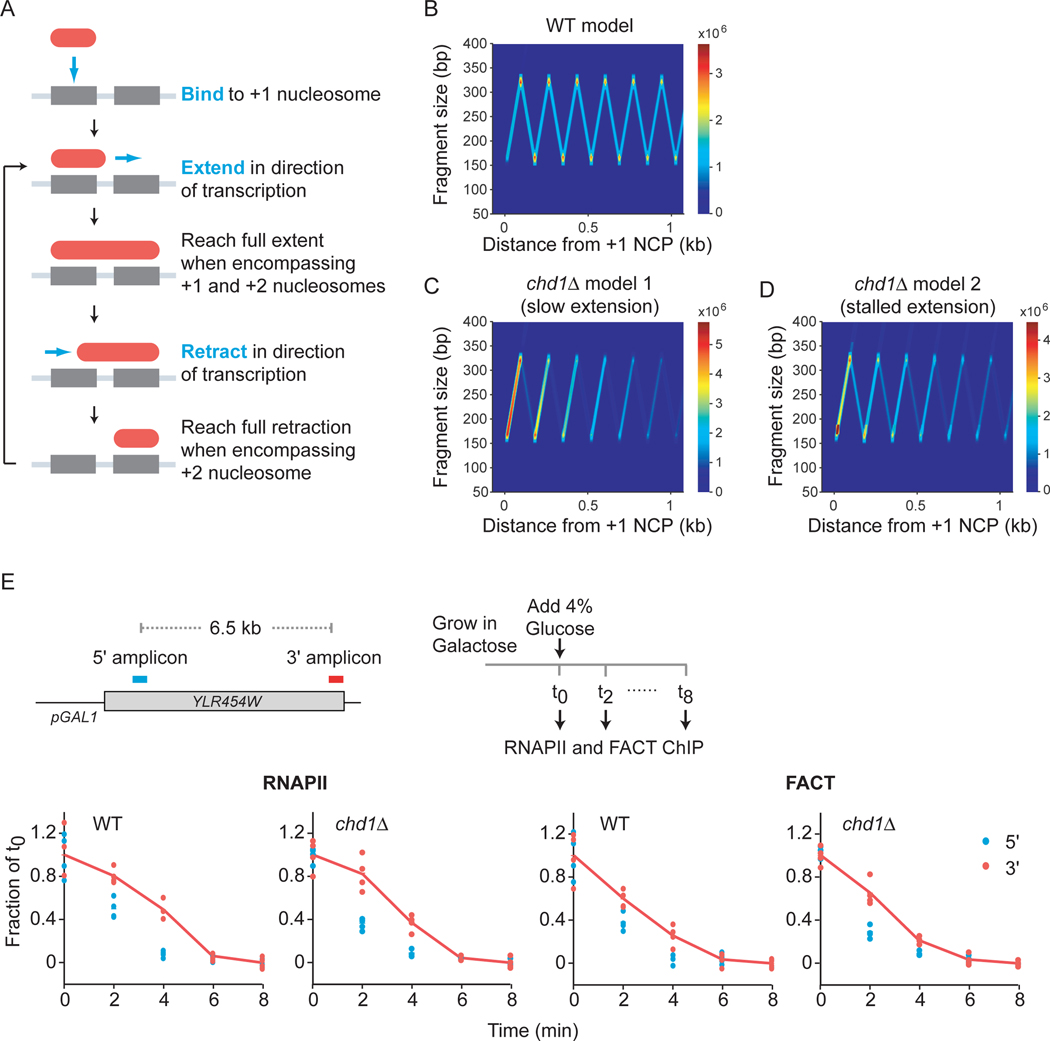
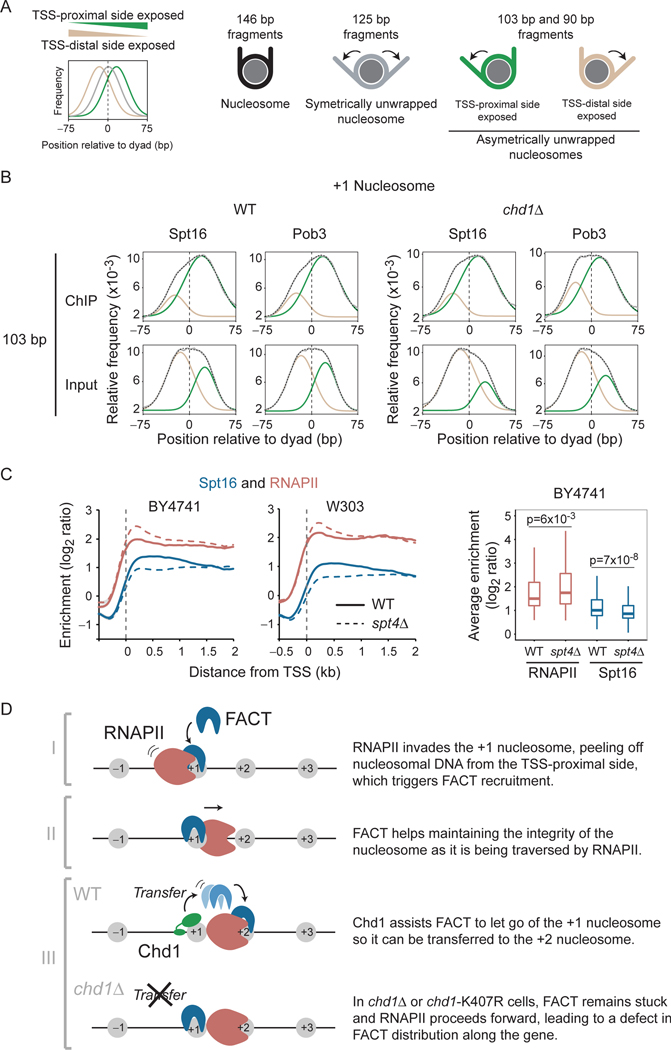
The histone chaperone FACT occupies transcribed regions where it plays prominent roles in maintaining chromatin integrity and preserving epigenetic information. How it is targeted to transcribed regions, however, remains unclear. Proposed models include docking on the RNA polymerase II (RNAPII) C-terminal domain (CTD), recruitment by elongation factors, recognition of modified histone tails, and binding partially disassembled nucleosomes. Here, we systematically test these and other scenarios in Saccharomyces cerevisiae and find that FACT binds transcribed chromatin, not RNAPII. Through a combination of high-resolution genome-wide mapping, single-molecule tracking, and mathematical modeling, we propose that FACT recognizes the +1 nucleosome, as it is partially unwrapped by the engaging RNAPII, and spreads to downstream nucleosomes aided by the chromatin remodeler Chd1. Our work clarifies how FACT interacts with genes, suggests a processive mechanism for FACT function, and provides a framework to further dissect the molecular mechanisms of transcription-coupled histone chaperoning.
Keywords: Chd1; FACT; Pob3; RNA polymerase II; Spt16; chromatin remodeling; histone chaperone; mathematical modeling; nucleosome unwrapping; single-molecule tracking.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Similar articles
-
Resolution of transcription-induced hexasome-nucleosome complexes by Chd1 and FACT.Mol Cell. 2024 Sep 19;84(18):3423-3437.e8. doi: 10.1016/j.molcel.2024.08.022. Epub 2024 Sep 12. Mol Cell. 2024. PMID: 39270644
-
Histone chaperones FACT and Spt6 prevent histone variants from turning into histone deviants.Bioessays. 2016 May;38(5):420-6. doi: 10.1002/bies.201500122. Epub 2016 Mar 15. Bioessays. 2016. PMID: 26990181
-
The Chaperone FACT and Histone H2B Ubiquitination Maintain S. pombe Genome Architecture through Genic and Subtelomeric Functions.Mol Cell. 2020 Feb 6;77(3):501-513.e7. doi: 10.1016/j.molcel.2019.11.016. Epub 2019 Dec 11. Mol Cell. 2020. PMID: 31837996 Free PMC article.
-
The role of FACT in managing chromatin: disruption, assembly, or repair?Nucleic Acids Res. 2020 Dec 2;48(21):11929-11941. doi: 10.1093/nar/gkaa912. Nucleic Acids Res. 2020. PMID: 33104782 Free PMC article. Review.
-
Beyond Chaperoning: The Multifaceted Role of FACT in Chromatin Transactions.Int J Mol Sci. 2025 May 28;26(11):5176. doi: 10.3390/ijms26115176. Int J Mol Sci. 2025. PMID: 40507984 Free PMC article. Review.
Cited by
-
Pluripotency state transition of embryonic stem cells requires the turnover of histone chaperone FACT on chromatin.iScience. 2023 Nov 28;27(1):108537. doi: 10.1016/j.isci.2023.108537. eCollection 2024 Jan 19. iScience. 2023. PMID: 38213626 Free PMC article.
-
Resolution of transcription-induced hexasome-nucleosome complexes by Chd1 and FACT.Mol Cell. 2024 Sep 19;84(18):3423-3437.e8. doi: 10.1016/j.molcel.2024.08.022. Epub 2024 Sep 12. Mol Cell. 2024. PMID: 39270644
-
Structural insights into nuclear transcription by eukaryotic DNA-dependent RNA polymerases.Nat Rev Mol Cell Biol. 2022 Sep;23(9):603-622. doi: 10.1038/s41580-022-00476-9. Epub 2022 May 3. Nat Rev Mol Cell Biol. 2022. PMID: 35505252 Review.
-
Structural Transition of the Nucleosome during Transcription Elongation.Cells. 2023 May 14;12(10):1388. doi: 10.3390/cells12101388. Cells. 2023. PMID: 37408222 Free PMC article. Review.
-
A direct interaction between the Chd1 CHCT domain and Rtf1 controls Chd1 distribution and nucleosome positioning on active genes.bioRxiv [Preprint]. 2024 Dec 6:2024.12.06.627179. doi: 10.1101/2024.12.06.627179. bioRxiv. 2024. PMID: 39677735 Free PMC article. Preprint.
References
-
- Bataille AR, Jeronimo C, Jacques PE, Laramee L, Fortin ME, Forest A, Bergeron M, Hanes SD, and Robert F. (2012). A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol Cell 45, 158–170. - PubMed
-
- Beati P, and Chereji RV (2020). Creating 2D Occupancy Plots Using plot2DO. Methods Mol Biol 2117, 93–108. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
