

GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis
- PMID: 34381026
- PMCID: PMC8357841
- DOI: 10.1038/s41467-021-24859-2
GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis
Abstract
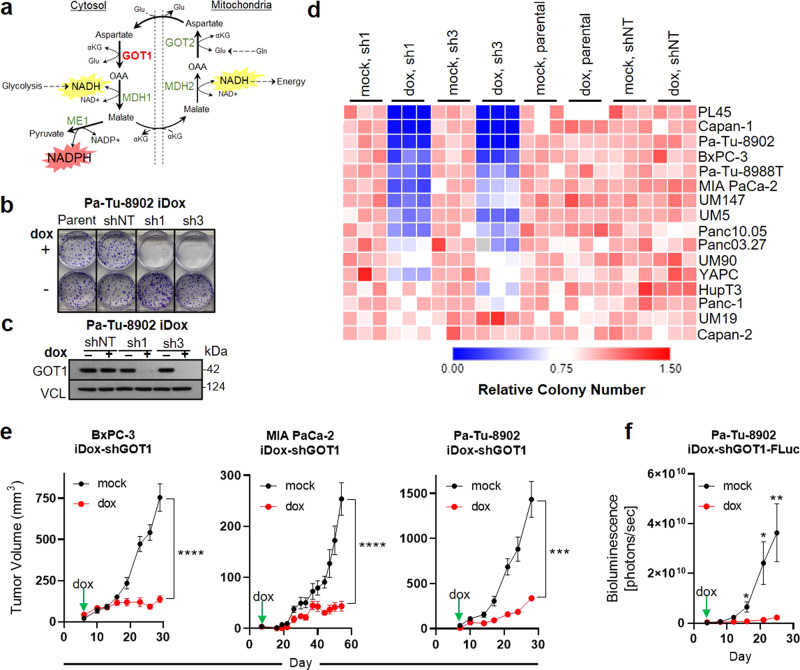
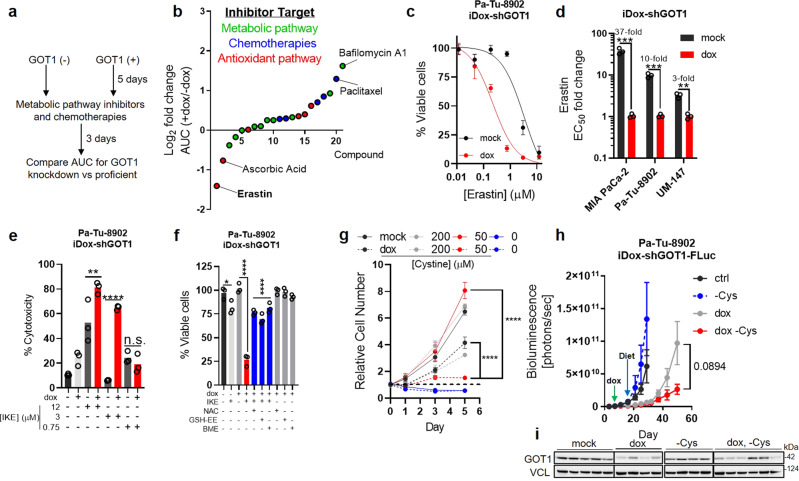
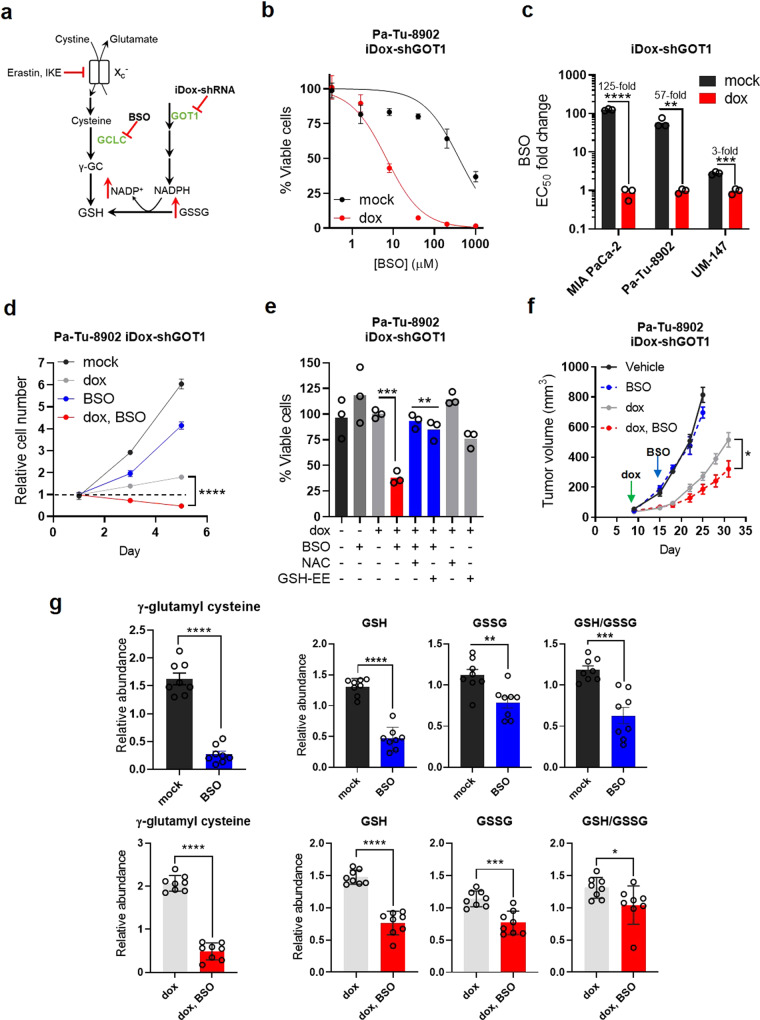
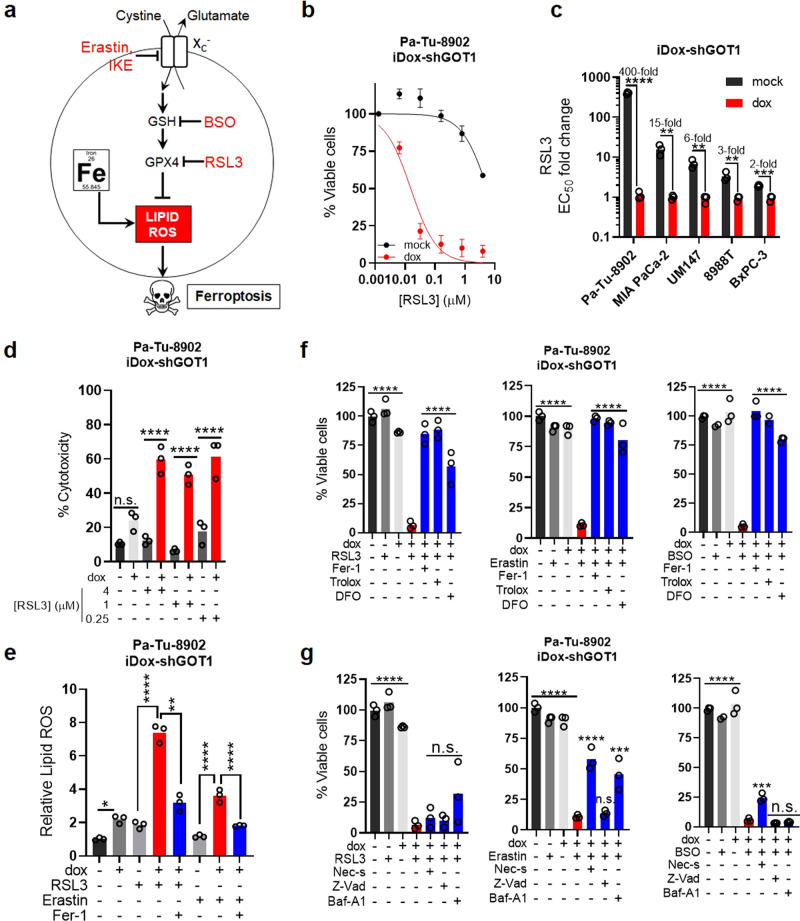
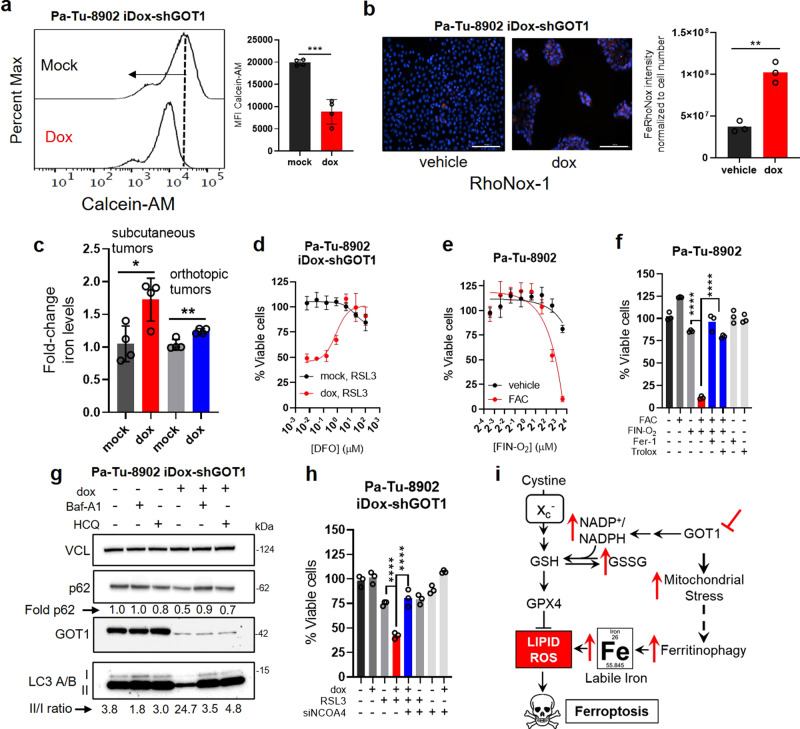
Cancer metabolism is rewired to support cell survival in response to intrinsic and environmental stressors. Identification of strategies to target these adaptions is an area of active research. We previously described a cytosolic aspartate aminotransaminase (GOT1)-driven pathway in pancreatic cancer used to maintain redox balance. Here, we sought to identify metabolic dependencies following GOT1 inhibition to exploit this feature of pancreatic cancer and to provide additional insight into regulation of redox metabolism. Using pharmacological methods, we identify cysteine, glutathione, and lipid antioxidant function as metabolic vulnerabilities following GOT1 withdrawal. We demonstrate that targeting any of these pathways triggers ferroptosis, an oxidative, iron-dependent form of cell death, in GOT1 knockdown cells. Mechanistically, we reveal that GOT1 inhibition represses mitochondrial metabolism and promotes a catabolic state. Consequently, we find that this enhances labile iron availability through autophagy, which potentiates the activity of ferroptotic stimuli. Overall, our study identifies a biochemical connection between GOT1, iron regulation, and ferroptosis.
© 2021. The Author(s).
Conflict of interest statement
C.A.L. has received consulting fees from Astellas Pharmaceuticals and is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting the GOT1-pathway as a therapeutic approach: (1) US Patent No: 20190136238, 05/09/2019; International Patent No: WO2013177426-A2, 04/23/2015 “Targeting the glutamine to pyruvate pathway for treatment of oncogenic Kras-associated cancer”, and (2) US Patent No: 2015126580-A1, 05/07/2015 “Methods for diagnosing and treating oncogenic Kras-associated cancer”. The other authors declare no competing interests.
Figures
References
-
- Perera RM, Bardeesy N. Pancreatic cancer metabolism: breaking it down to build it back up. Cancer Discov. 2015;5:1247–1261. doi: 10.1158/2159-8290.CD-15-0671. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA148828/CA/NCI NIH HHS/United States
- R01 DK095201/DK/NIDDK NIH HHS/United States
- R01 CA198074/CA/NCI NIH HHS/United States
- P30 CA046592/CA/NCI NIH HHS/United States
- R01 CA248160/CA/NCI NIH HHS/United States
- U01 CA224145/CA/NCI NIH HHS/United States
- T32 CA140044/CA/NCI NIH HHS/United States
- R01 CA215607/CA/NCI NIH HHS/United States
- R01 CA245546/CA/NCI NIH HHS/United States
- K22 CA237752/CA/NCI NIH HHS/United States
- P30 DK089503/DK/NIDDK NIH HHS/United States
- R01 CA151588/CA/NCI NIH HHS/United States
- T32 CA009676/CA/NCI NIH HHS/United States
- R01 CA244931/CA/NCI NIH HHS/United States
- P30 DK034933/DK/NIDDK NIH HHS/United States
- T32 DK094775/DK/NIDDK NIH HHS/United States
- R37 CA237421/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
