

Cycling cancer persister cells arise from lineages with distinct programs
- PMID: 34381210
- PMCID: PMC9209846
- DOI: 10.1038/s41586-021-03796-6
Cycling cancer persister cells arise from lineages with distinct programs
Abstract
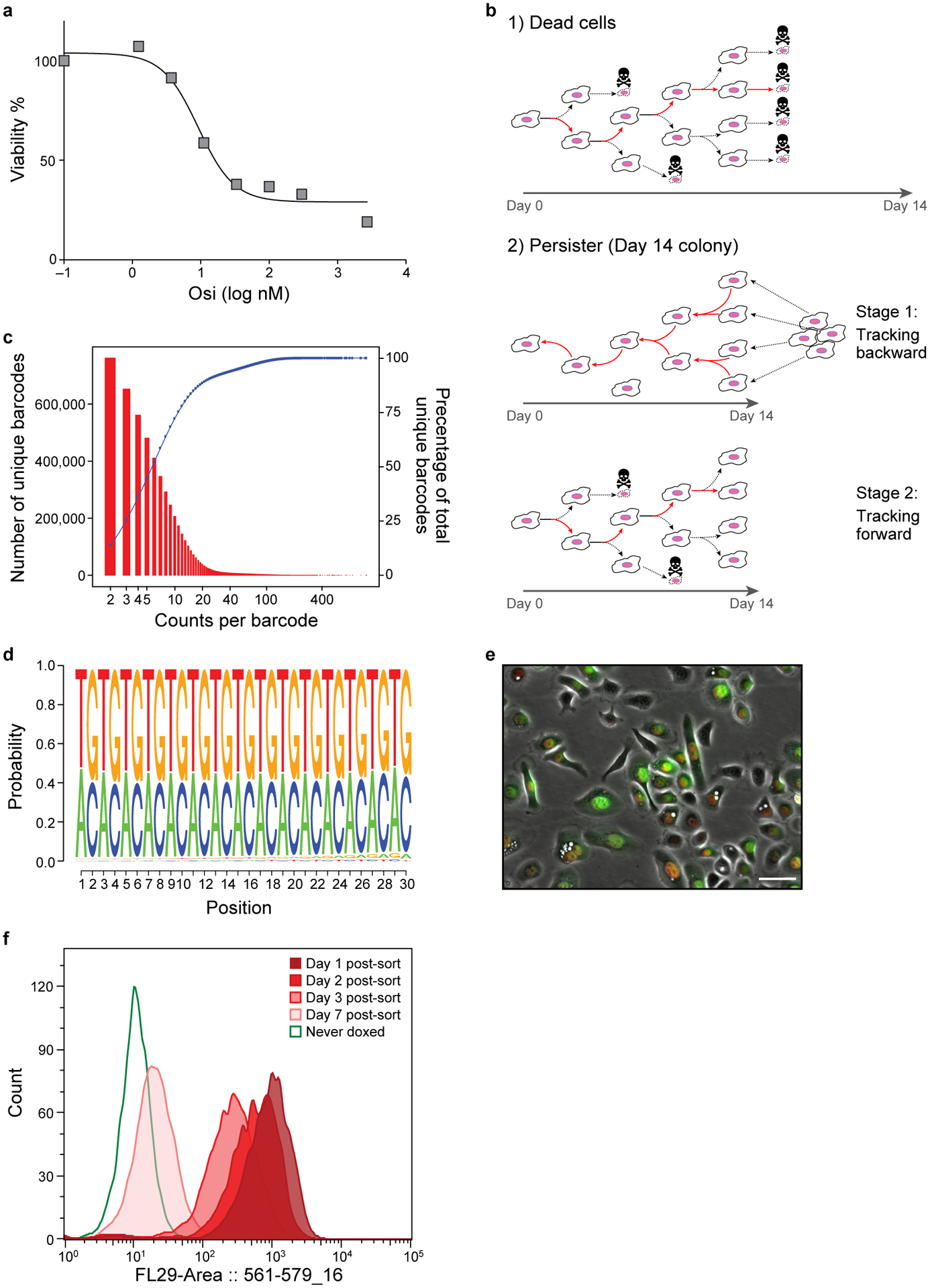
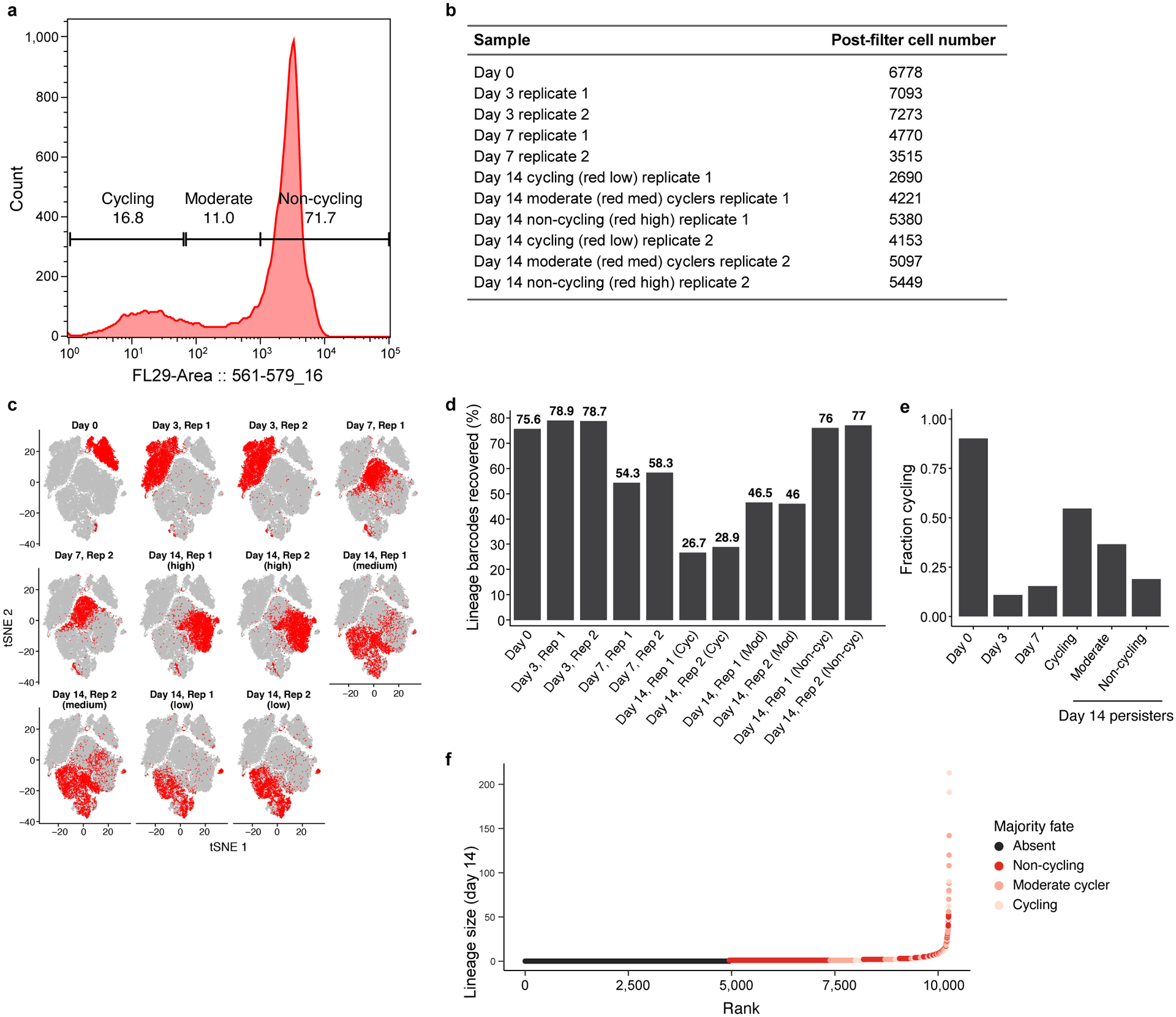
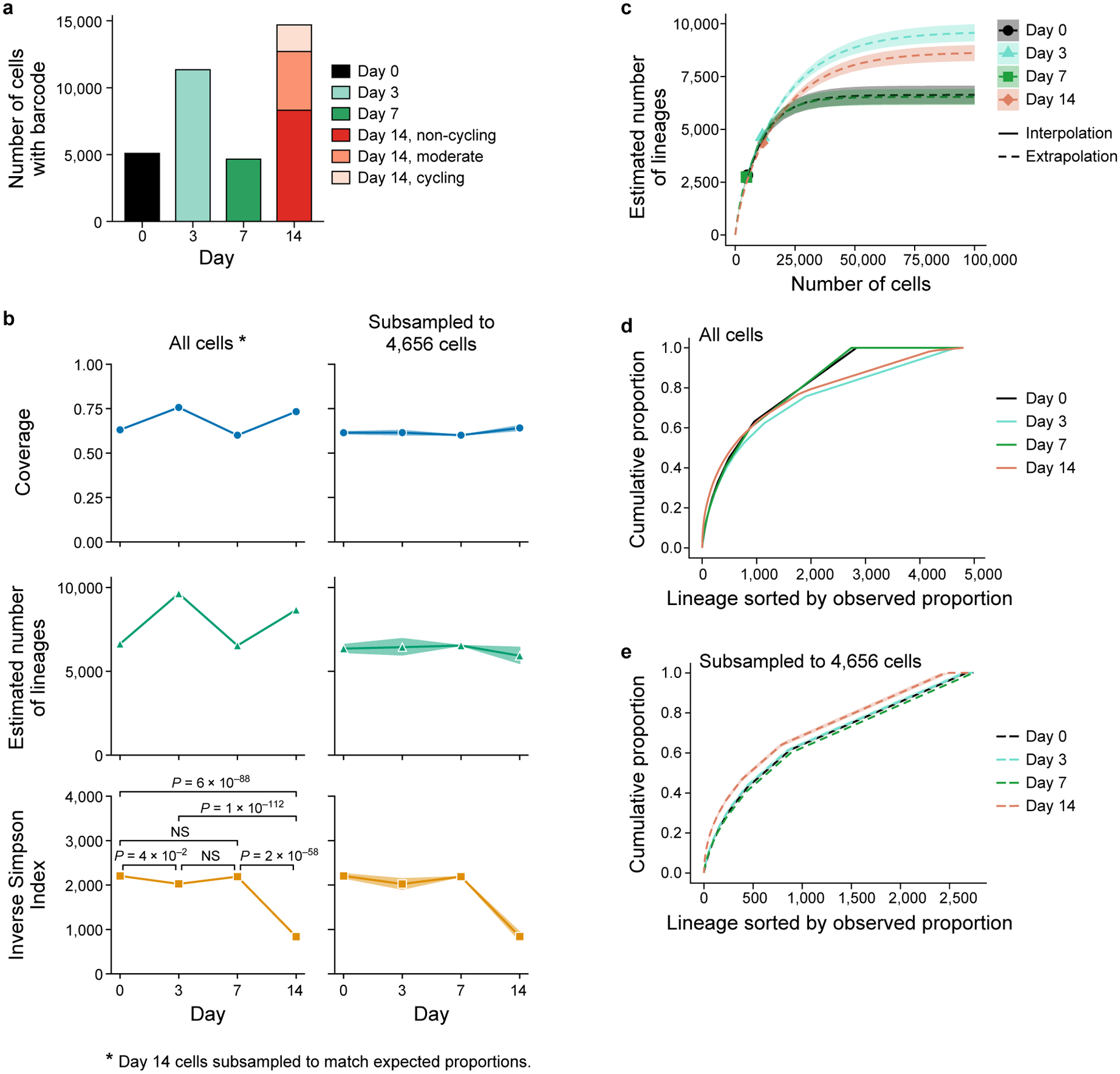
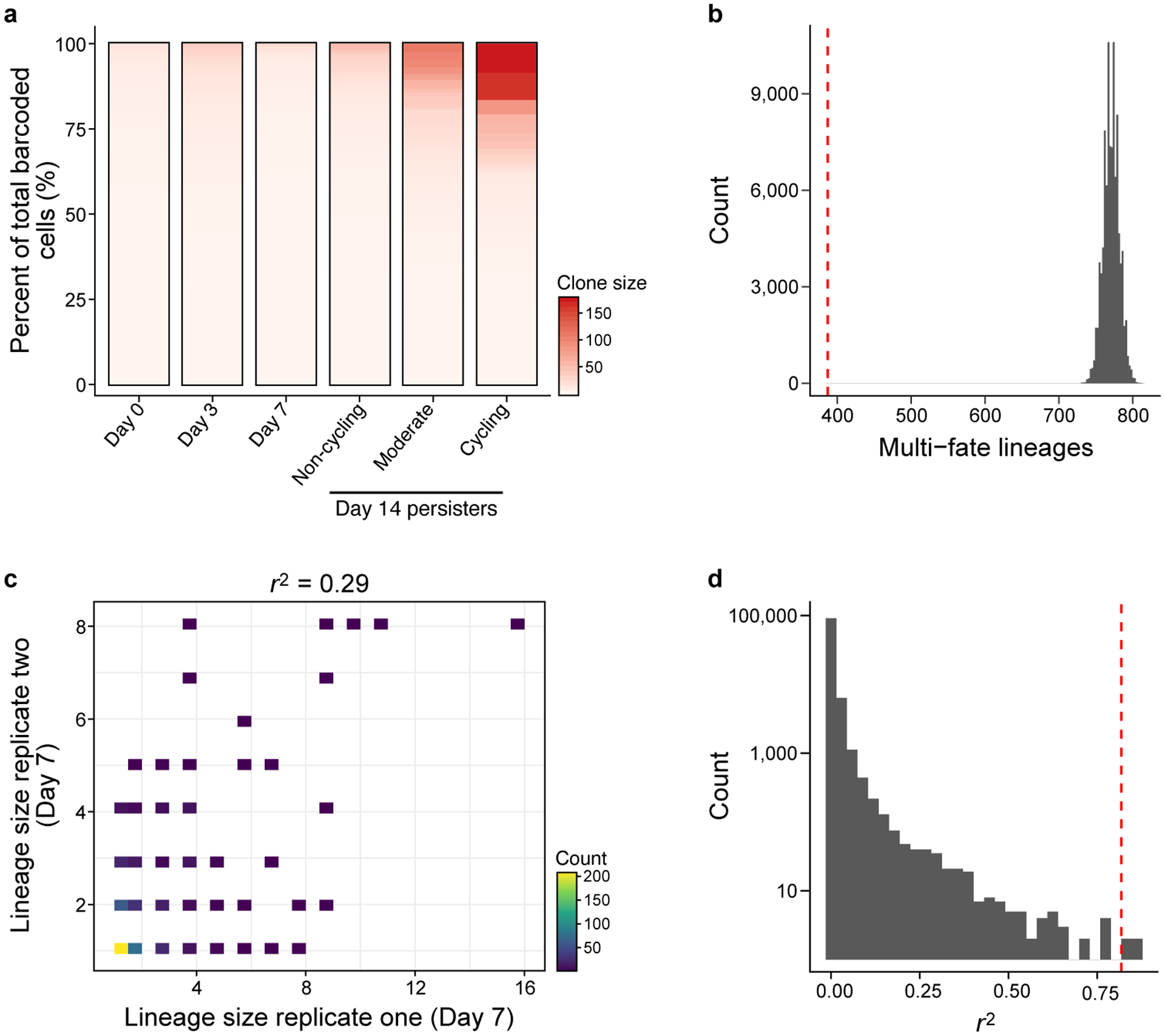
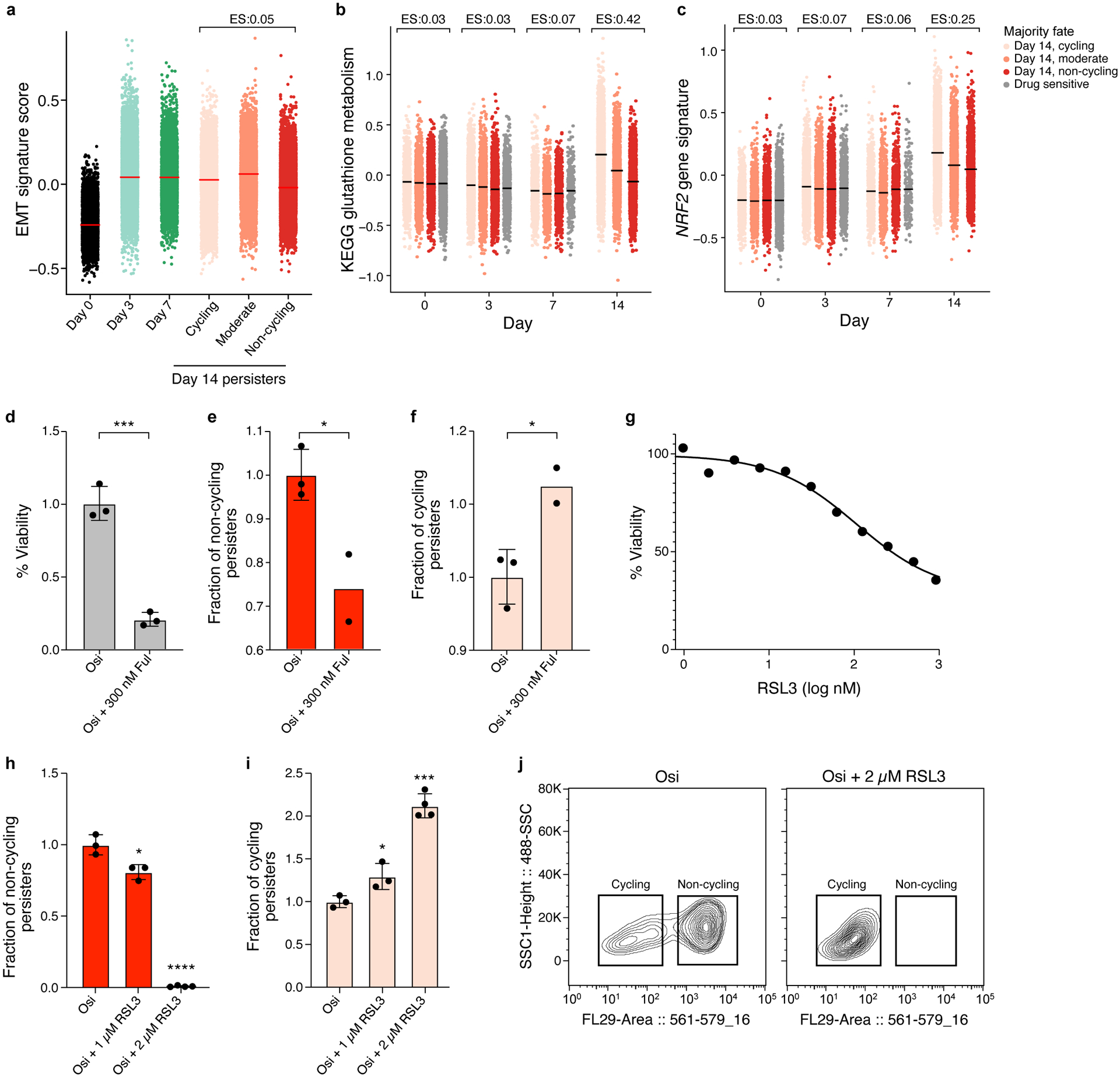
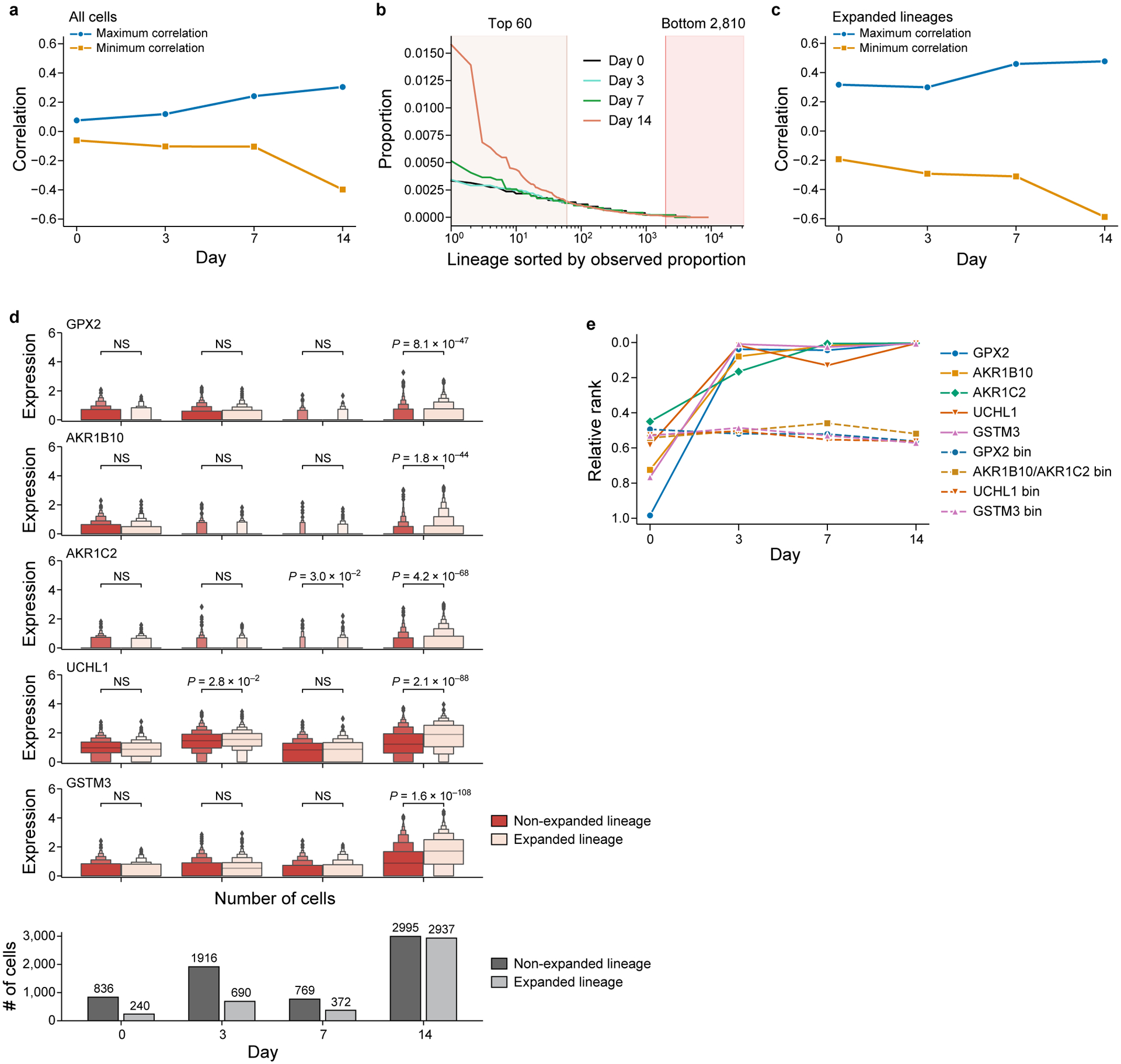
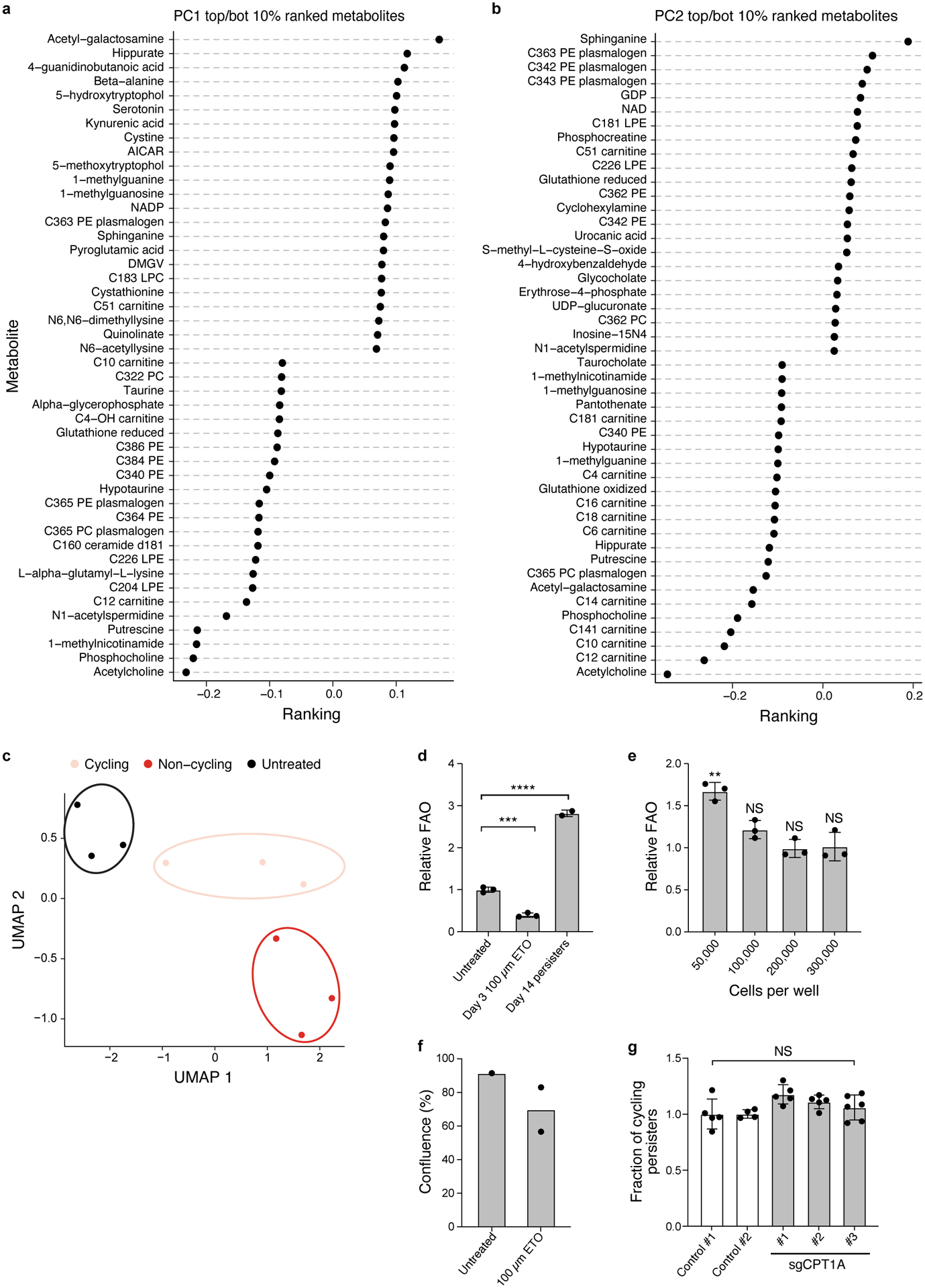
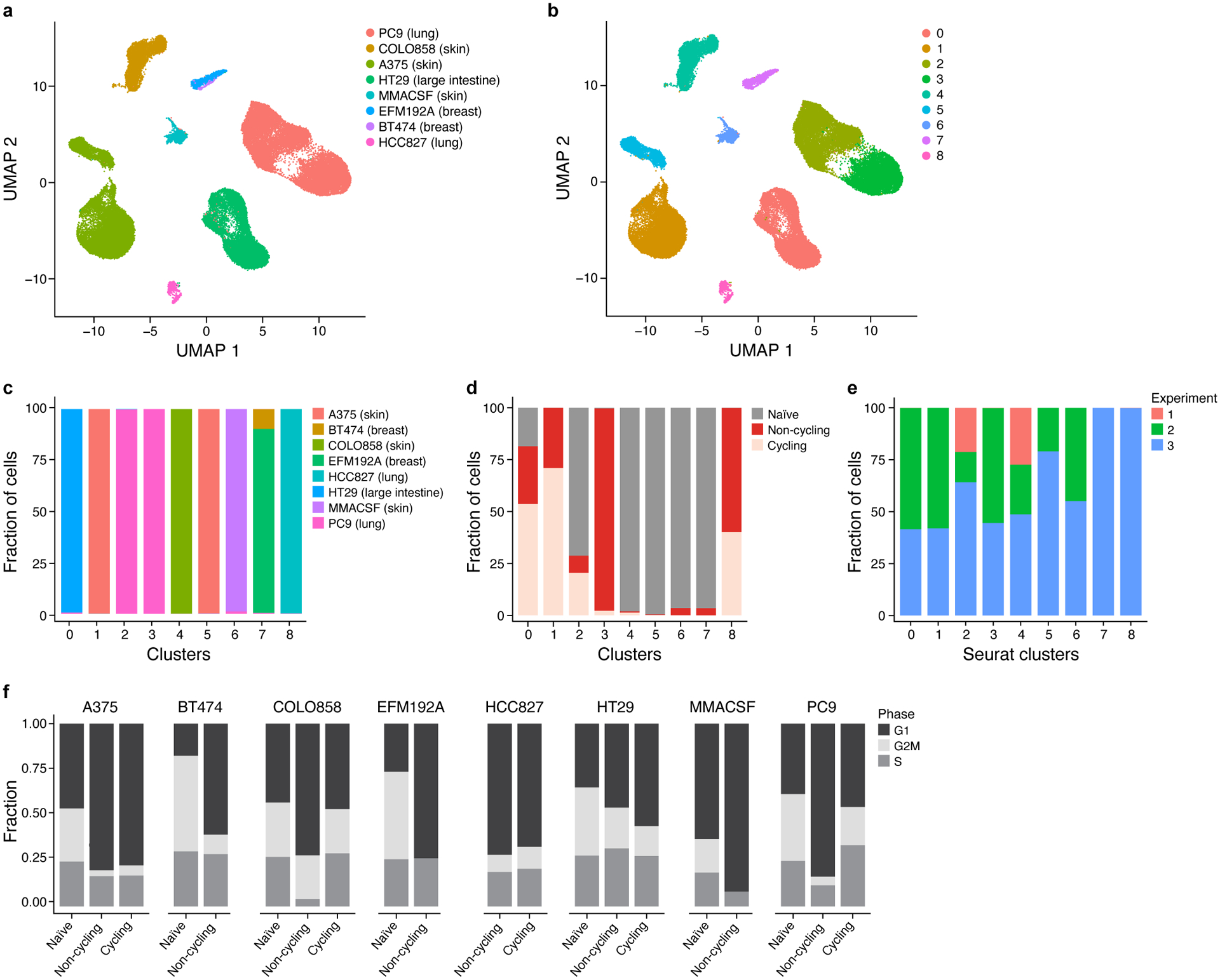
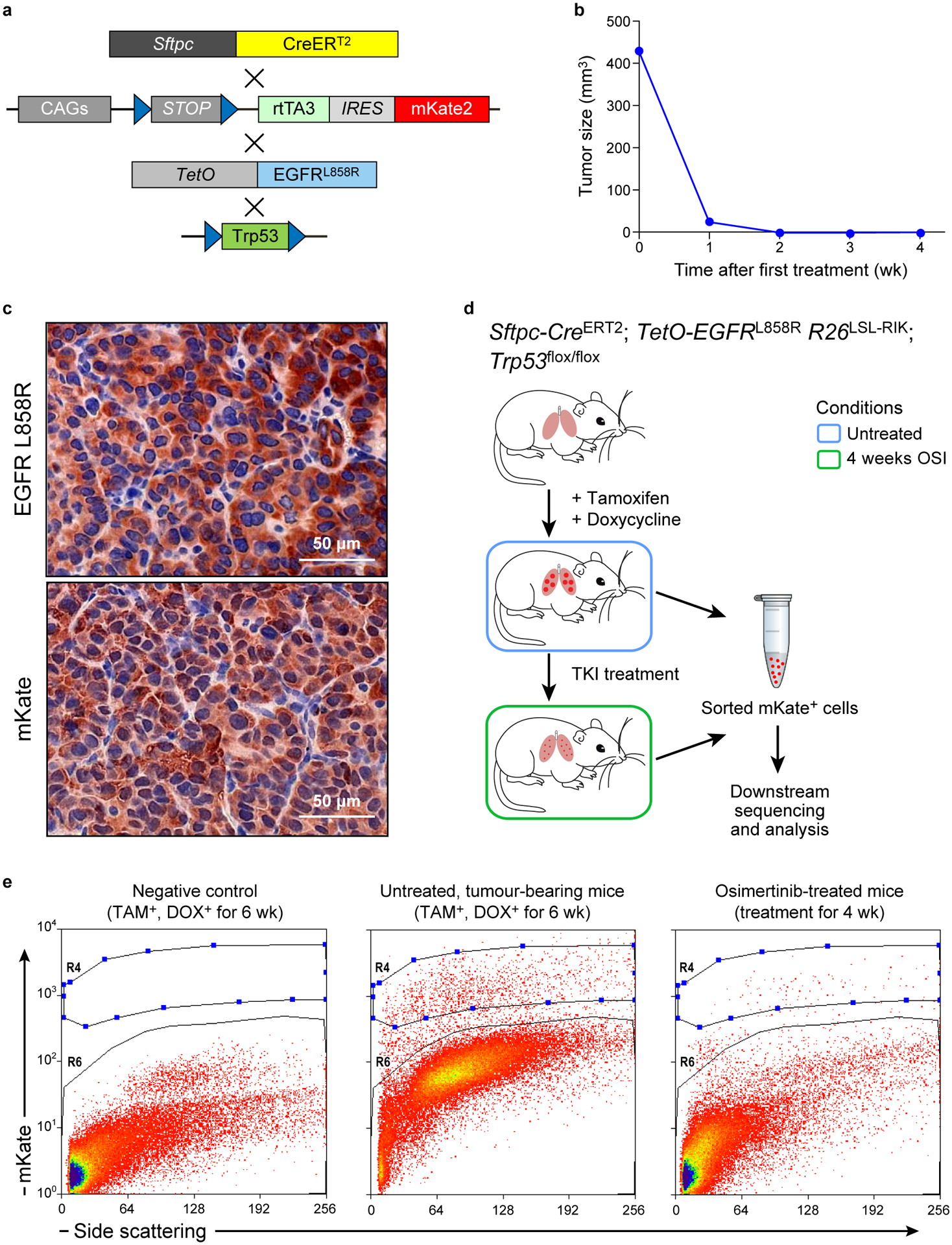
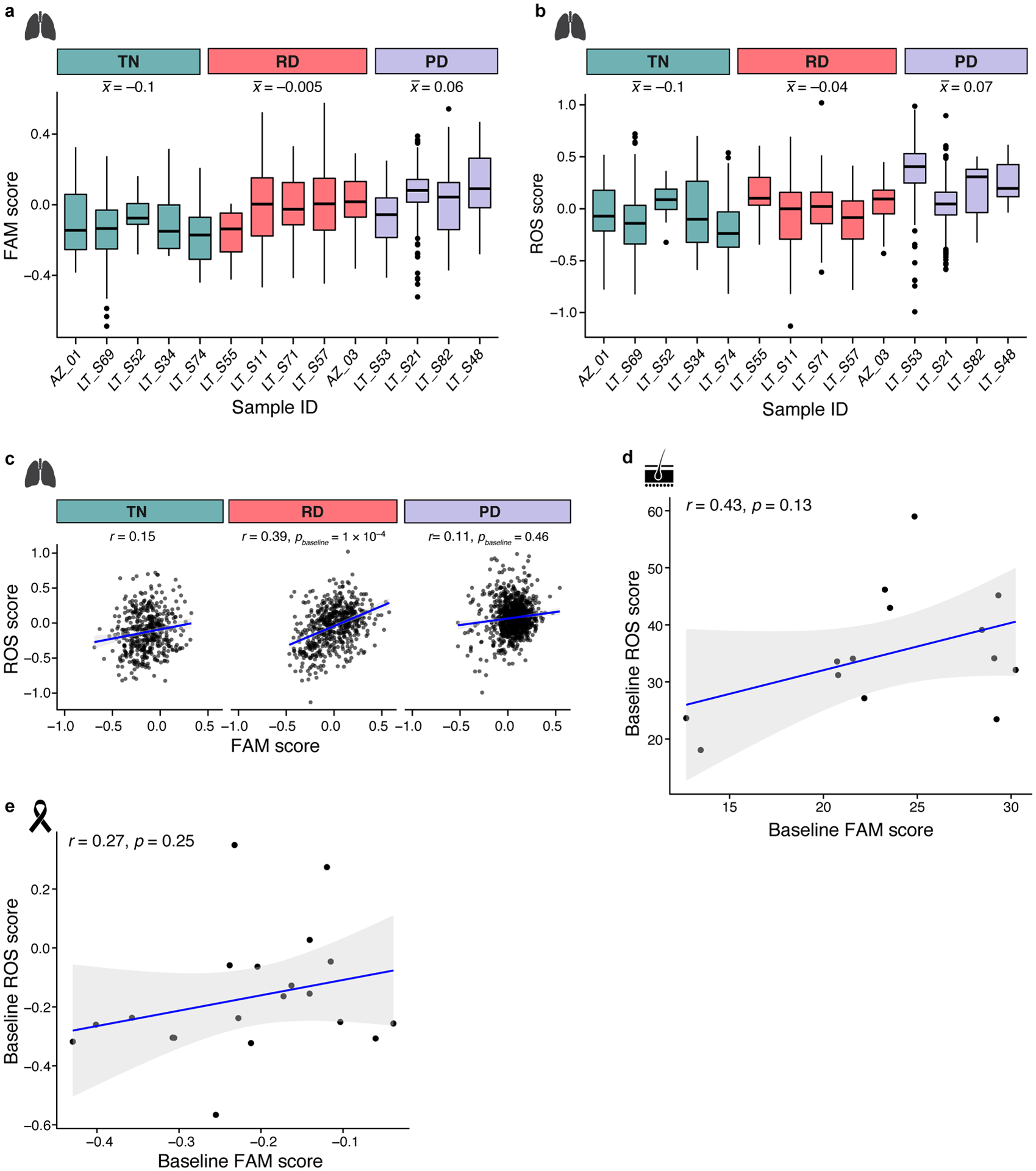
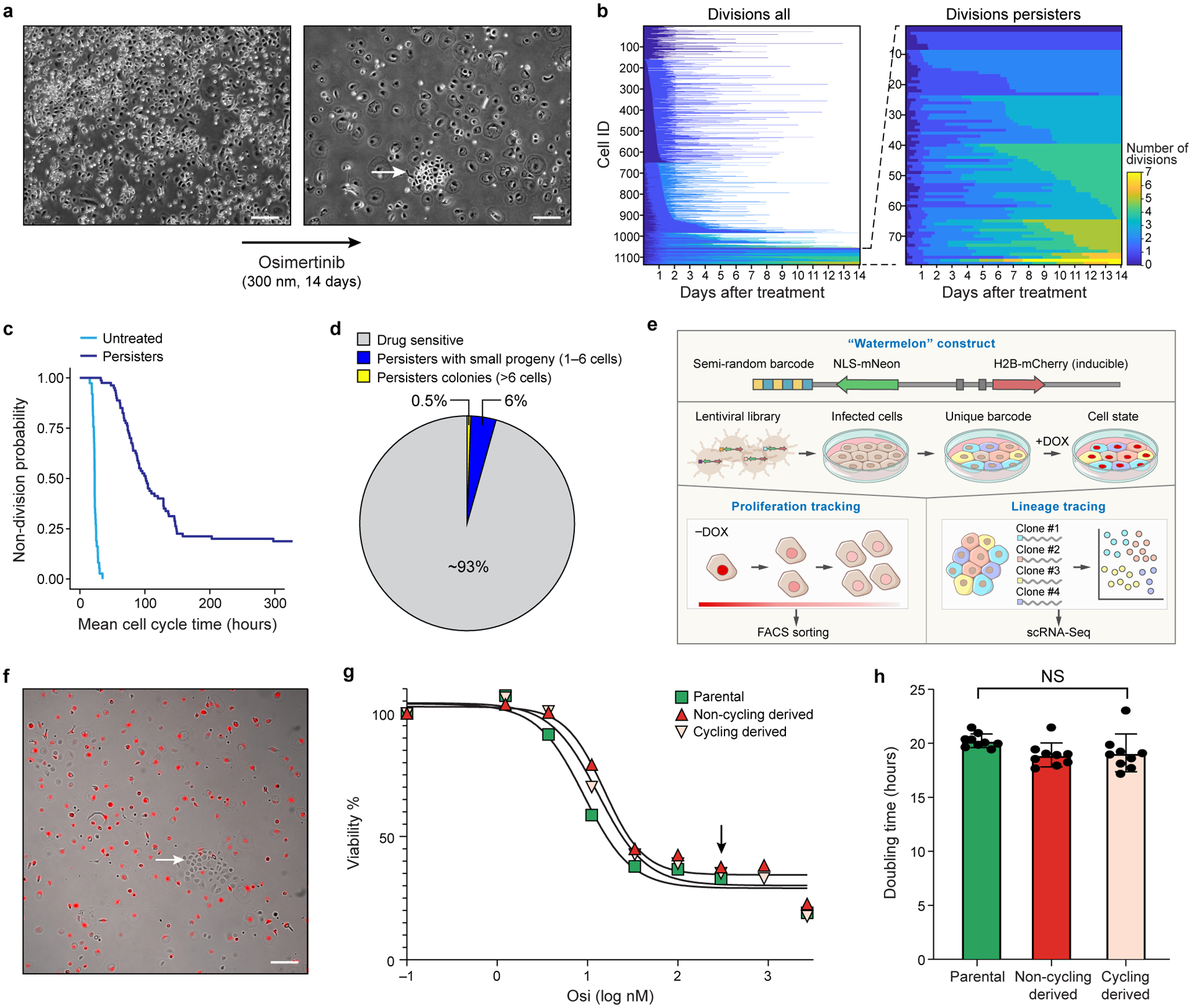
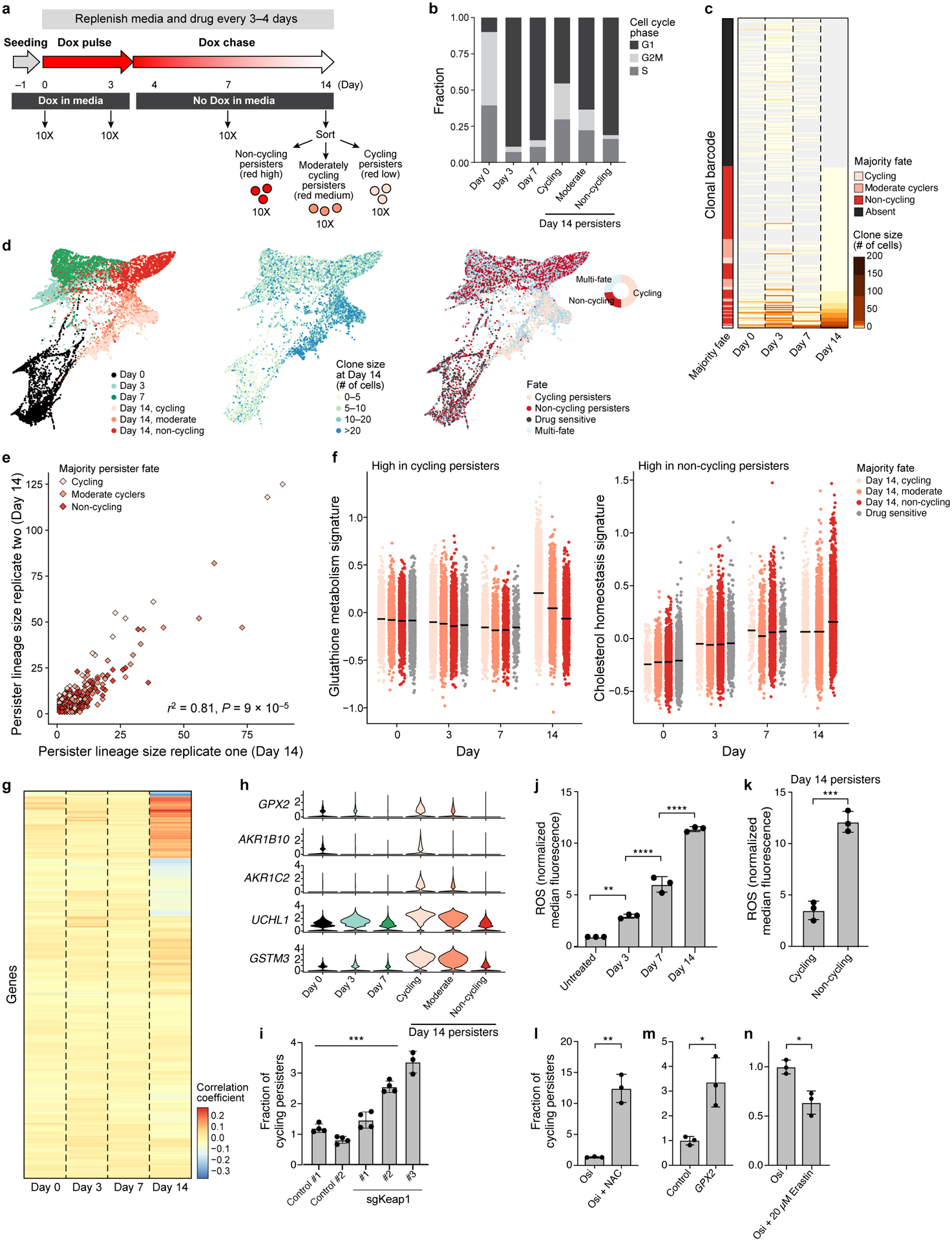
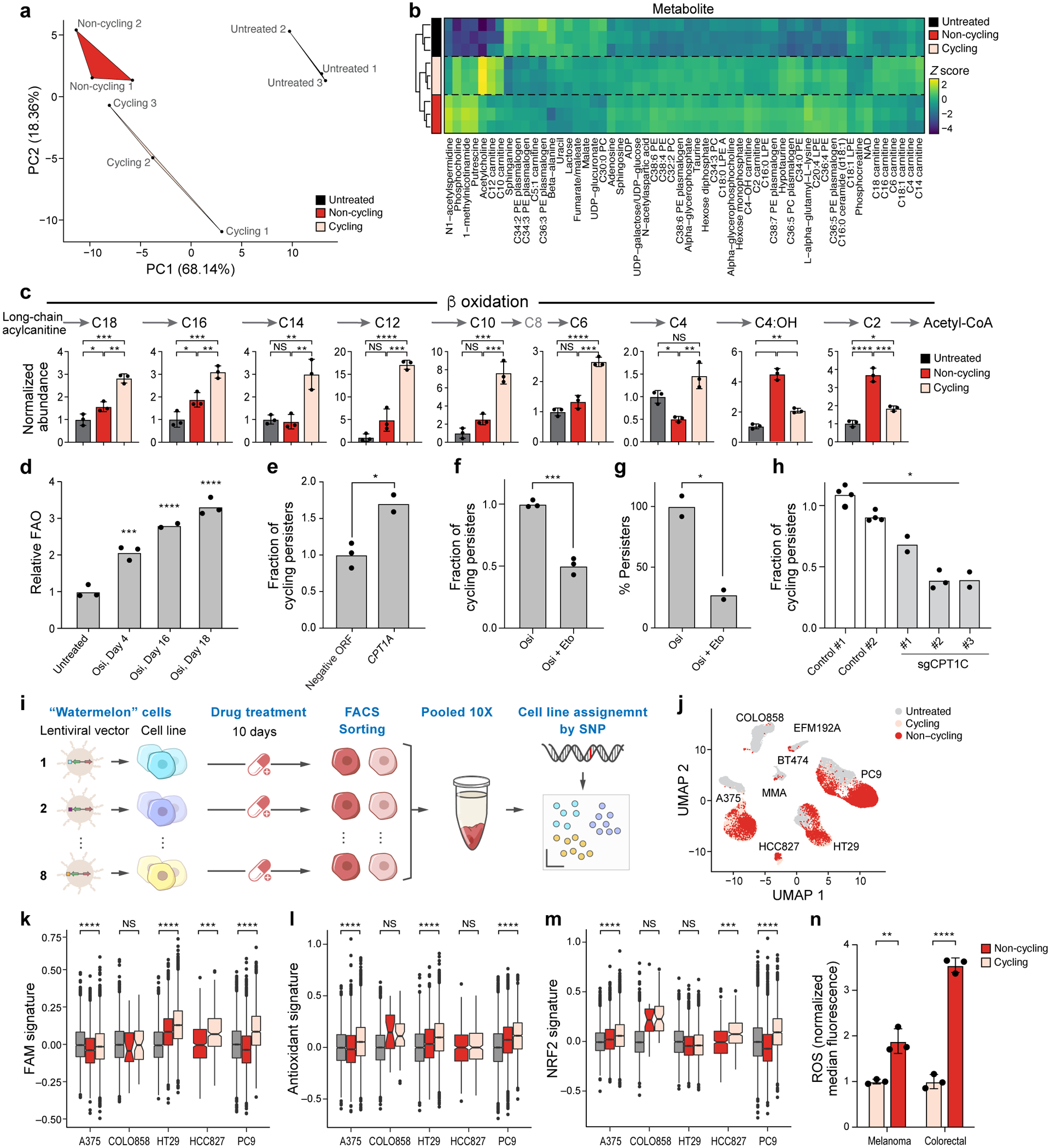
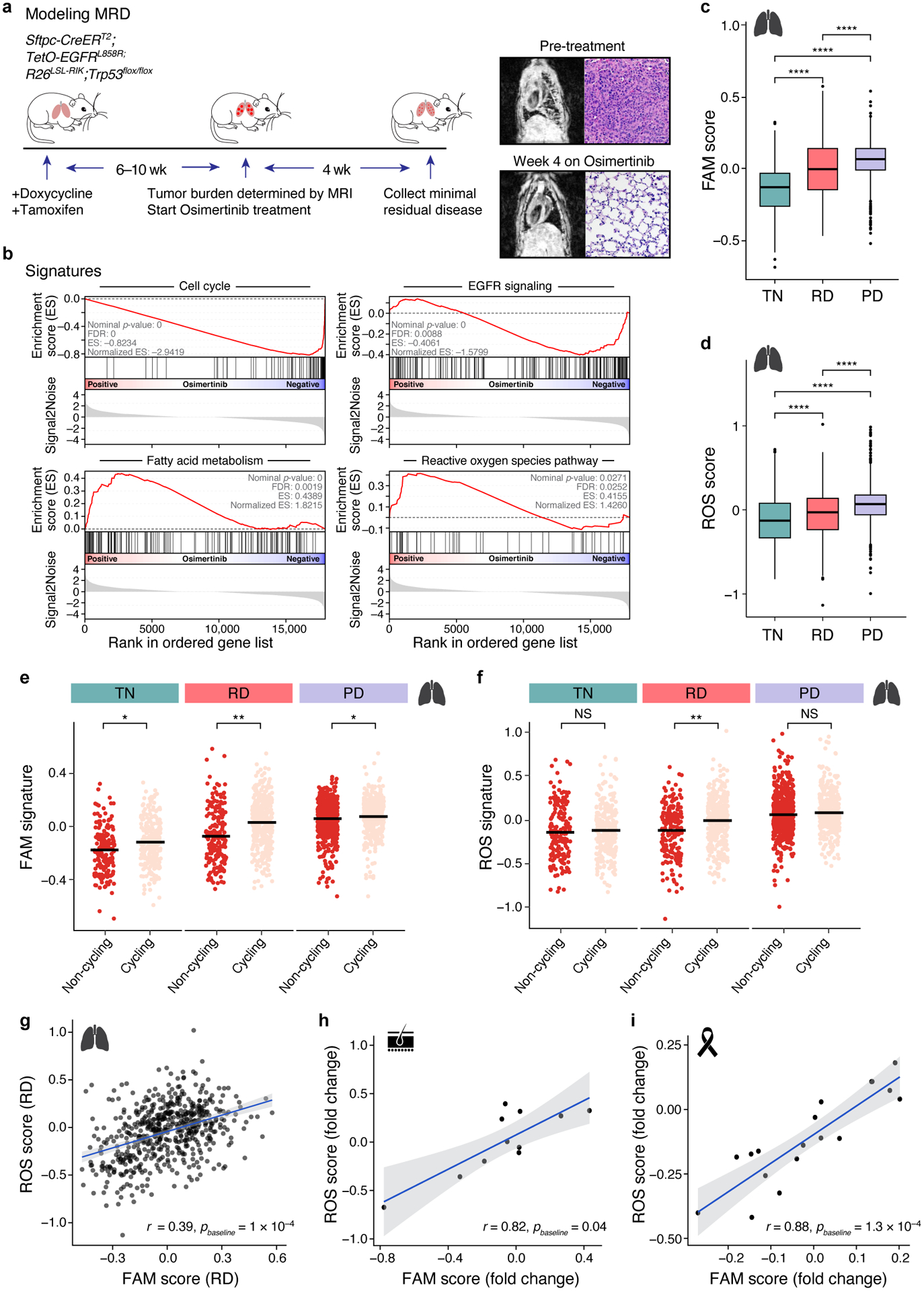
Non-genetic mechanisms have recently emerged as important drivers of cancer therapy failure1, where some cancer cells can enter a reversible drug-tolerant persister state in response to treatment2. Although most cancer persisters remain arrested in the presence of the drug, a rare subset can re-enter the cell cycle under constitutive drug treatment. Little is known about the non-genetic mechanisms that enable cancer persisters to maintain proliferative capacity in the presence of drugs. To study this rare, transiently resistant, proliferative persister population, we developed Watermelon, a high-complexity expressed barcode lentiviral library for simultaneous tracing of each cell's clonal origin and proliferative and transcriptional states. Here we show that cycling and non-cycling persisters arise from different cell lineages with distinct transcriptional and metabolic programs. Upregulation of antioxidant gene programs and a metabolic shift to fatty acid oxidation are associated with persister proliferative capacity across multiple cancer types. Impeding oxidative stress or metabolic reprogramming alters the fraction of cycling persisters. In human tumours, programs associated with cycling persisters are induced in minimal residual disease in response to multiple targeted therapies. The Watermelon system enabled the identification of rare persister lineages that are preferentially poised to proliferate under drug pressure, thus exposing new vulnerabilities that can be targeted to delay or even prevent disease recurrence.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
Comment in
-
A persistent look at how tumours evade therapy.Nature. 2021 Aug;596(7873):491-493. doi: 10.1038/d41586-021-02117-1. Nature. 2021. PMID: 34381221 No abstract available.
-
Cycling persister cells.Nat Rev Cancer. 2021 Nov;21(11):683. doi: 10.1038/s41568-021-00409-2. Nat Rev Cancer. 2021. PMID: 34522021 No abstract available.
-
Acquired cancer tyrosine kinase inhibitor resistance: ROS as critical determinants.Signal Transduct Target Ther. 2021 Dec 24;6(1):437. doi: 10.1038/s41392-021-00844-5. Signal Transduct Target Ther. 2021. PMID: 34952897 Free PMC article. No abstract available.
-
Hunting down rare drug-tolerant cycling cells with Watermelon.Nat Rev Cancer. 2022 Aug;22(8):434-435. doi: 10.1038/s41568-022-00483-0. Nat Rev Cancer. 2022. PMID: 35538369 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
