

Natural history, outcome measures and trial readiness in LAMA2-related muscular dystrophy and SELENON-related myopathy in children and adults: protocol of the LAST STRONG study
- PMID: 34384384
- PMCID: PMC8357962
- DOI: 10.1186/s12883-021-02336-z
Natural history, outcome measures and trial readiness in LAMA2-related muscular dystrophy and SELENON-related myopathy in children and adults: protocol of the LAST STRONG study
Abstract
Background: SELENON (SEPN1)-related myopathy (SELENON-RM) is a rare congenital myopathy characterized by slowly progressive proximal muscle weakness, early onset spine rigidity and respiratory insufficiency. A muscular dystrophy caused by mutations in the LAMA2 gene (LAMA2-related muscular dystrophy, LAMA2-MD) has a similar clinical phenotype, with either a severe, early-onset due to complete Laminin subunit α2 deficiency (merosin-deficient congenital muscular dystrophy type 1A (MDC1A)), or a mild, childhood- or adult-onset due to partial Laminin subunit α2 deficiency. For both muscle diseases, no curative treatment options exist, yet promising preclinical studies are ongoing. Currently, there is a paucity on natural history data and appropriate clinical and functional outcome measures are needed to reach trial readiness.
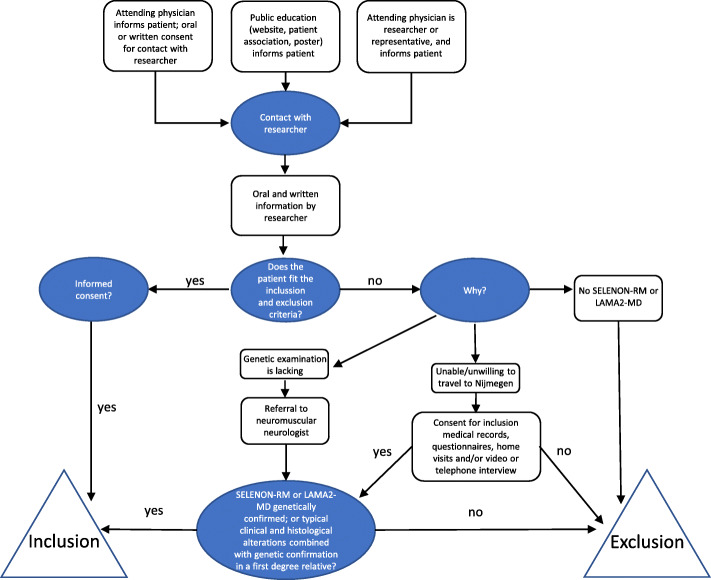
Methods: LAST STRONG is a natural history study in Dutch-speaking patients of all ages diagnosed with SELENON-RM or LAMA2-MD, starting August 2020. Patients have four visits at our hospital over a period of 1.5 year. At all visits, they undergo standardized neurological examination, hand-held dynamometry (age ≥ 5 years), functional measurements, questionnaires (patient report and/or parent proxy; age ≥ 2 years), muscle ultrasound including diaphragm, pulmonary function tests (spirometry, maximal inspiratory and expiratory pressure, sniff nasal inspiratory pressure; age ≥ 5 years), and accelerometry for 8 days (age ≥ 2 years); at visit one and three, they undergo cardiac evaluation (electrocardiogram, echocardiography; age ≥ 2 years), spine X-ray (age ≥ 2 years), dual-energy X-ray absorptiometry (DEXA-)scan (age ≥ 2 years) and full body magnetic resonance imaging (MRI) (age ≥ 10 years). All examinations are adapted to the patient's age and functional abilities. Correlation between key parameters within and between subsequent visits will be assessed.
Discussion: Our study will describe the natural history of patients diagnosed with SELENON-RM or LAMA2-MD, enabling us to select relevant clinical and functional outcome measures for reaching clinical trial-readiness. Moreover, our detailed description (deep phenotyping) of the clinical features will optimize clinical management and will establish a well-characterized baseline cohort for prospective follow-up.
Conclusion: Our natural history study is an essential step for reaching trial readiness in SELENON-RM and LAMA2-MD.
Trial registration: This study has been approved by medical ethical reviewing committee Region Arnhem-Nijmegen (NL64269.091.17, 2017-3911) and is registered at ClinicalTrial.gov ( NCT04478981 ).
Keywords: All ages; LAMA2; Laminin subunit α2 deficiency; Merosin-deficient congenital muscular dystrophy type 1A (MDC1A); Natural history; Outcome measures; SELENON; SEPN1; Trial readiness.
© 2021. The Author(s).
Conflict of interest statement
Professor Jan A.M. Smeitink is Chief Executive Officer of Khondrion. All other authors declare that they have no competing interests.
Figures
References
-
- Villar-Quiles RN, von der Hagen M, Métay C, Gonzalez V, Donkervoort S, Bertini E, Castiglioni C, Chaigne D, Colomer J, Cuadrado ML, de Visser M, Desguerre I, Eymard B, Goemans N, Kaindl A, Lagrue E, Lütschg J, Malfatti E, Mayer M, Merlini L, Orlikowski D, Reuner U, Salih MA, Schlotter-Weigel B, Stoetter M, Straub V, Topaloglu H, Urtizberea JA, van der Kooi A, Wilichowski E, Romero NB, Fardeau M, Bönnemann CG, Estournet B, Richard P, Quijano-Roy S, Schara U, Ferreiro A. The clinical, histologic, and genotypic spectrum of. Neurology. 2020;95(11):e1512–e1e27. doi: 10.1212/WNL.0000000000010327. - DOI - PMC - PubMed
-
- Geranmayeh F, Clement E, Feng LH, Sewry C, Pagan J, Mein R, Abbs S, Brueton L, Childs AM, Jungbluth H, de Goede CG, Lynch B, Lin JP, Chow G, Sousa C, O’Mahony O, Majumdar A, Straub V, Bushby K, Muntoni F. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. 2010;20(4):241–250. doi: 10.1016/j.nmd.2010.02.001. - DOI - PubMed
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
