

Population sequencing data reveal a compendium of mutational processes in the human germ line
- PMID: 34385354
- PMCID: PMC9217108
- DOI: 10.1126/science.aba7408
Population sequencing data reveal a compendium of mutational processes in the human germ line
Abstract
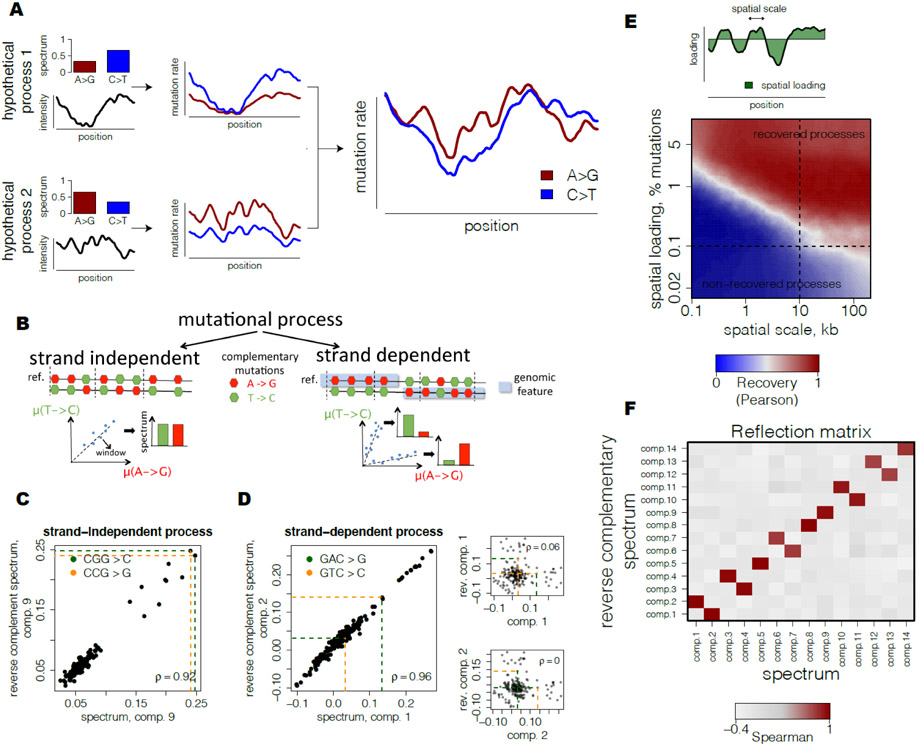
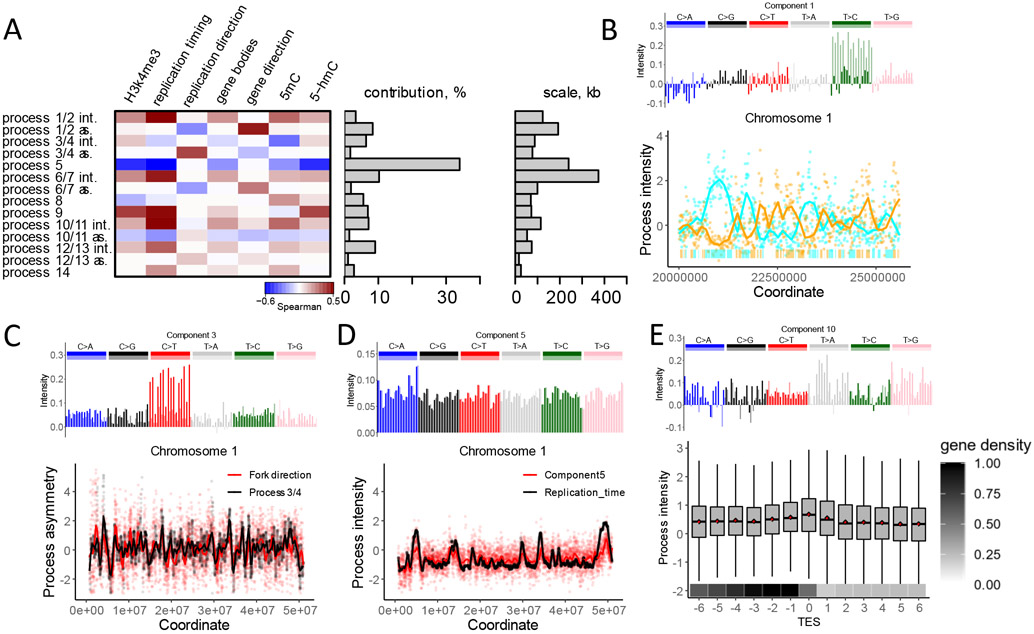
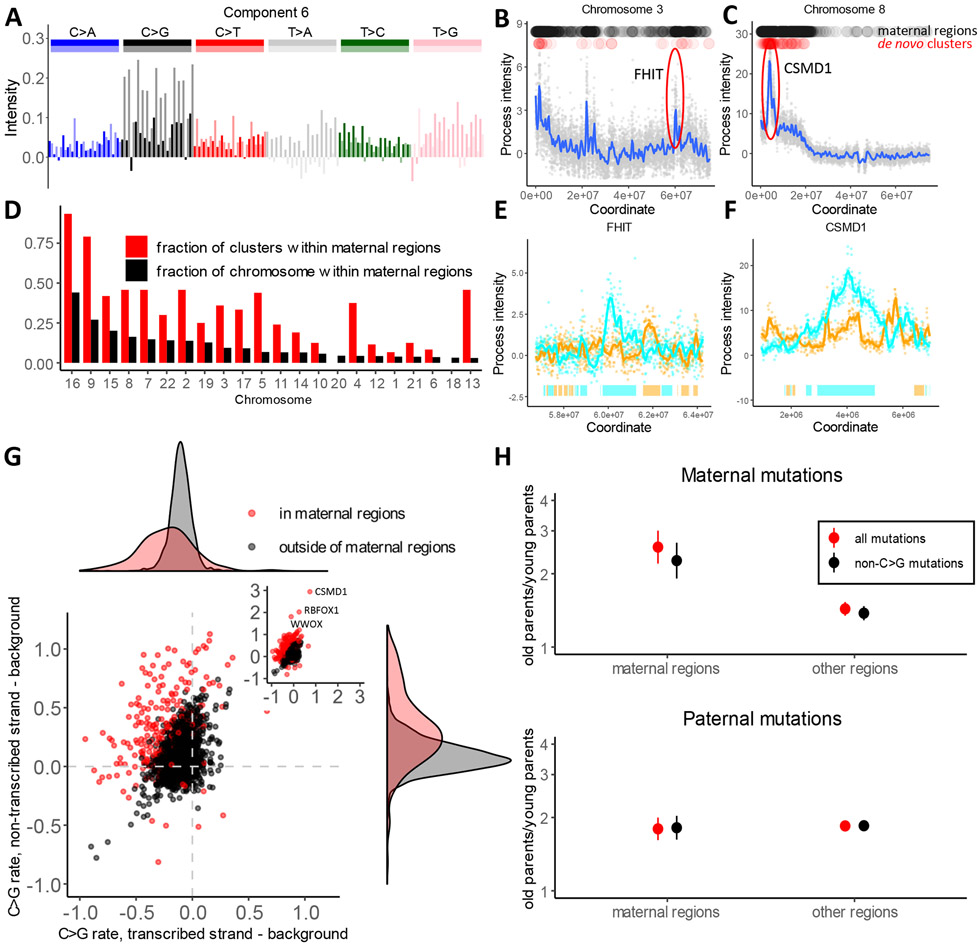
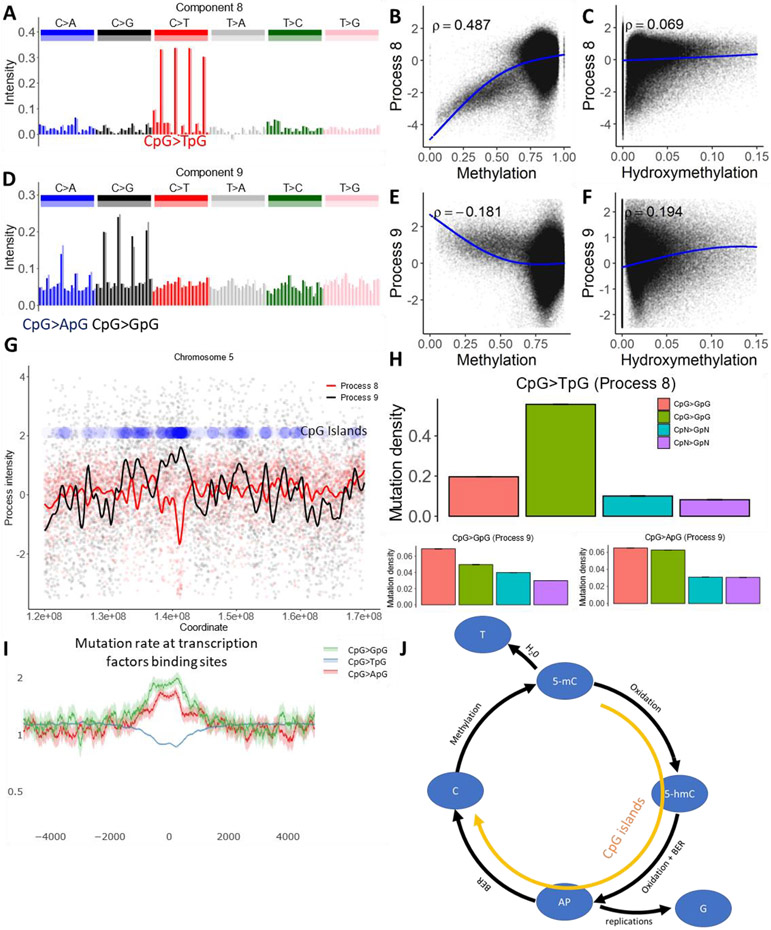
Biological mechanisms underlying human germline mutations remain largely unknown. We statistically decompose variation in the rate and spectra of mutations along the genome using volume-regularized nonnegative matrix factorization. The analysis of a sequencing dataset (TOPMed) reveals nine processes that explain the variation in mutation properties between loci. We provide a biological interpretation for seven of these processes. We associate one process with bulky DNA lesions that are resolved asymmetrically with respect to transcription and replication. Two processes track direction of replication fork and replication timing, respectively. We identify a mutagenic effect of active demethylation primarily acting in regulatory regions and a mutagenic effect of long interspersed nuclear elements. We localize a mutagenic process specific to oocytes from population sequencing data. This process appears transcriptionally asymmetric.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
References
-
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JHJ, Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol 15, 465–481 (2014). - PubMed
-
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Imielinsk M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR, Signatures of mutational processes in human cancer. Nature. 500, 415–421 (2013). - PMC - PubMed
Material and methods references
-
- Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, Pitsillides AN, LeFaive J, Lee S, Tian X, Browning BL, Das S, Emde A-K, Clarke WE, Loesch DP, Shetty AC, Blackwell TW, Wong Q, Aguet F, Albert C, Alonso A, Ardlie KG, Aslibekyan S, Auer PL, Barnard J, Barr RG, Becker LC, Beer RL, Benjamin EJ, Bielak LF, Blangero J, Boehnke M, Bowden DW, Brody JA, Burchard EG, Cade BE, Casella JF, Chalazan B, Chen Y-DI, Cho MH, Choi SH, Chung MK, Clish CB, Correa A, Curran JE, Custer B, Darbar D, Daya M, de Andrade M, DeMeo DL, Dutcher SK, Ellinor PT, Emery LS, Fatkin D, Forer L, Fornage M, Franceschini N, Fuchsberger C, Fullerton SM, Germer S, Gladwin MT, Gottlieb DJ, Guo X, Hall ME, He J, Heard-Costa NL, Heckbert SR, Irvin MR, Johnsen JM, Johnson AD, Kardia SLR, Kelly T, Kelly S, Kenny EE, Kiel DP, Klemmer R, Konkle BA, Kooperberg C, Köttgen A, Lange LA, Lasky-Su J, Levy D, Lin X, Lin K-H, Liu C, Loos RJF, Garman L, Gerszten R, Lubitz SA, Lunetta KL, Mak ACY, Manichaikul A, Manning AK, Mathias RA, McManus DD, McGarvey ST, Meigs JB, Meyers DA, Mikulla JL, Minear MA, Mitchell B, Mohanty S, Montasser ME, Montgomery C, Morrison AC, Murabito JM, Natale A, Natarajan P, Nelson SC, North KE, O’Connell JR, Palmer ND, Pankratz N, Peloso GM, Peyser PA, Post WS, Psaty BM, Rao DC, Redline S, Reiner AP, Roden D, Rotter JI, Ruczinski I, Sarnowski C, Schoenherr S, Seo J-S, Seshadri S, Sheehan VA, Shoemaker MB, Smith AV, Smith NL, Smith JA, Sotoodehnia N, Stilp AM, Tang W, Taylor KD, Telen M, Thornton TA, Tracy RP, Berg DJVD, Vasan RS, Viaud-Martinez KA, Vrieze S, Weeks DE, Weir BS, Weiss ST, Weng L-C, Willer CJ, Zhang Y, Zhao X, Arnett DK, Ashley-Koch AE, Barnes KC, Boerwinkle E, Gabriel S, Gibbs R, Rice KM, Rich SS, Silverman E, Qasba P, Gan W, Topm. P. G. W. G. Trans-Omics for Precision Medicine (TOPMed) Program, Papanicolaou GJ, Nickerson DA, Browning SR, Zody MC, Zöllner S, Wilson JG, Cupples LA, Laurie CC, Jaquish CE, Hernandez RD, O’Connor TD, Abecasis GR, Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv, 563866 (2019). - PubMed
-
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation Consortium, Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536, 285–291 (2016). - PMC - PubMed
-
- Halldorsson BV, Palsson G, Stefansson OA, Jonsson H, Hardarson MT, Eggertsson HP, Gunnarsson B, Oddsson A, Halldorsson GH, Zink F, Gudjonsson SA, Frigge ML, Thorleifsson G, Sigurdsson A, Stacey SN, Sulem P, Masson G, Helgason A, Gudbjartsson DF, Thorsteinsdottir U, Stefansson K, Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science. 363, eaau1043 (2019). - PubMed
-
- An J-Y, Lin K, Zhu L, Werling DM, Dong S, Brand H, Wang HZ, Zhao X, Schwartz GB, Collins RL, Currall BB, Dastmalchi C, Dea J, Duhn C, Gilson MC, Klei L, Liang L, Markenscoff-Papadimitriou E, Pochareddy S, Ahituv N, Buxbaum JD, Coon H, Daly MJ, Kim YS, Marth GT, Neale BM, Quinlan AR, Rubenstein JL, Sestan N, State MW, Willsey AJ, Talkowski ME, Devlin B, Roeder K, Sanders SJ, Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science. 362, eaat6576 (2018). - PMC - PubMed
