

Patterns of within-host genetic diversity in SARS-CoV-2
- PMID: 34387545
- PMCID: PMC8363274
- DOI: 10.7554/eLife.66857
Patterns of within-host genetic diversity in SARS-CoV-2
Abstract
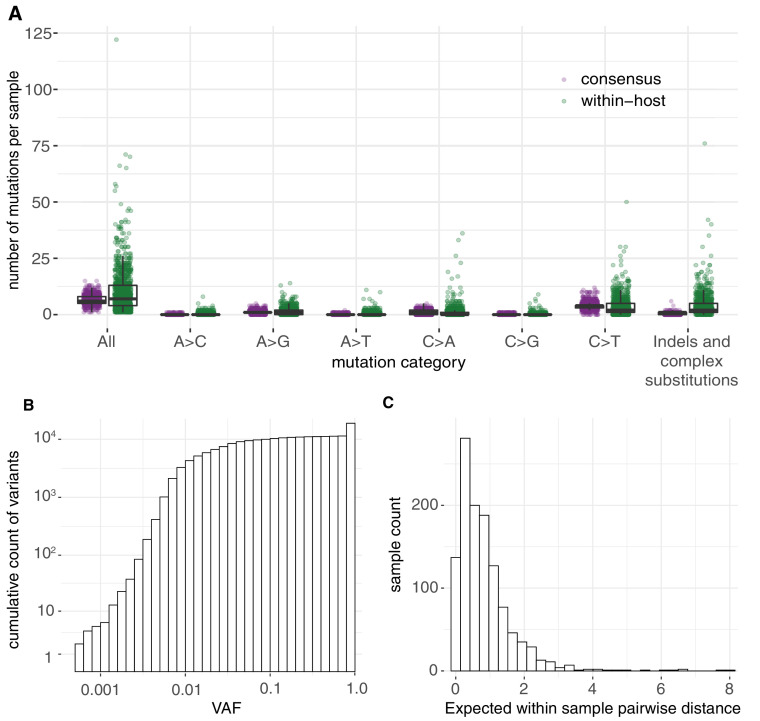
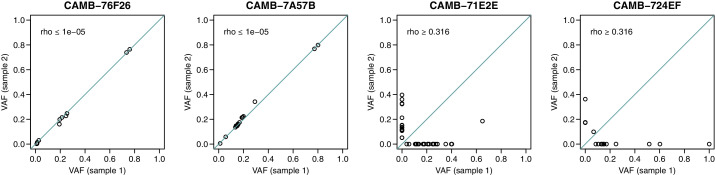
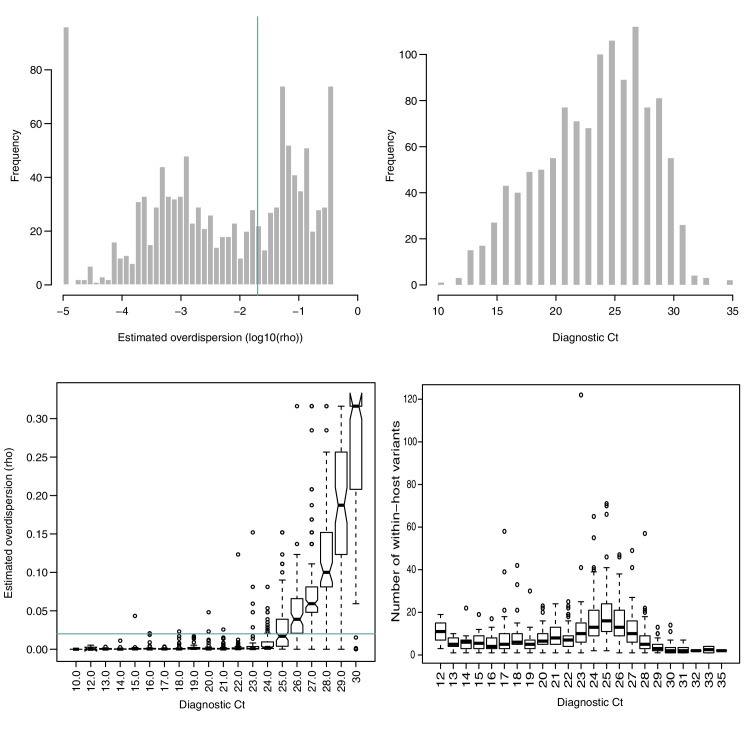
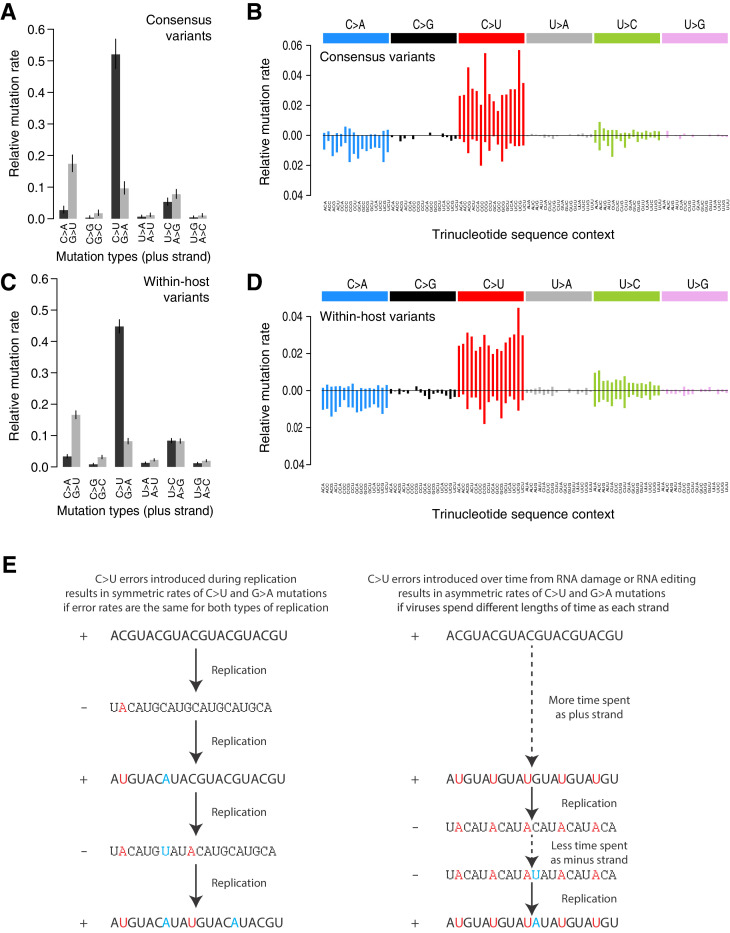
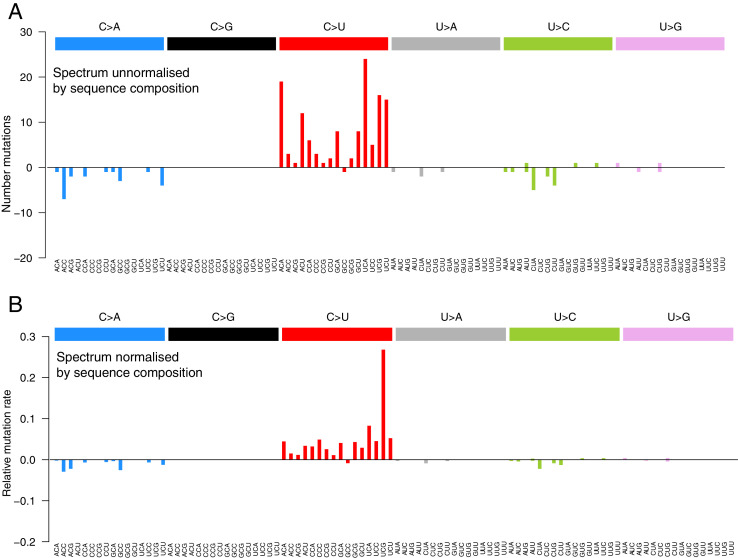
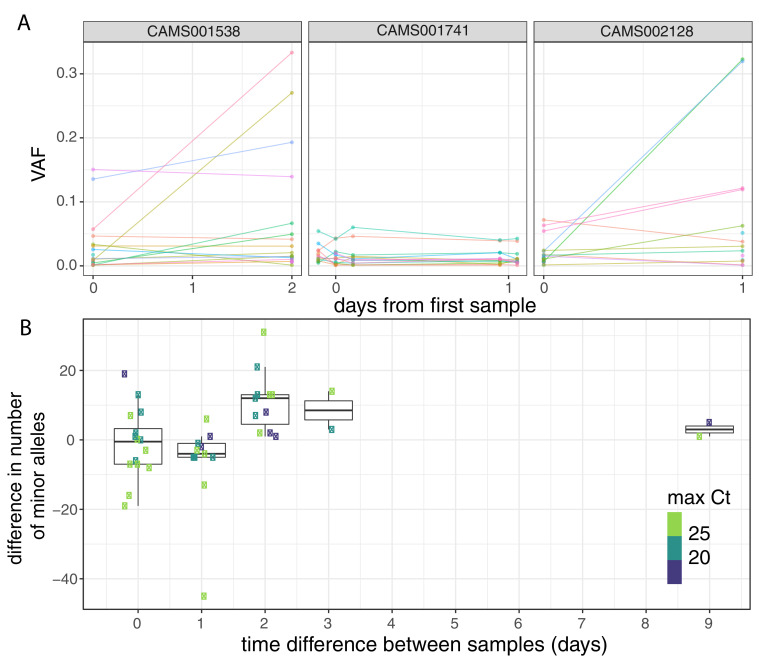
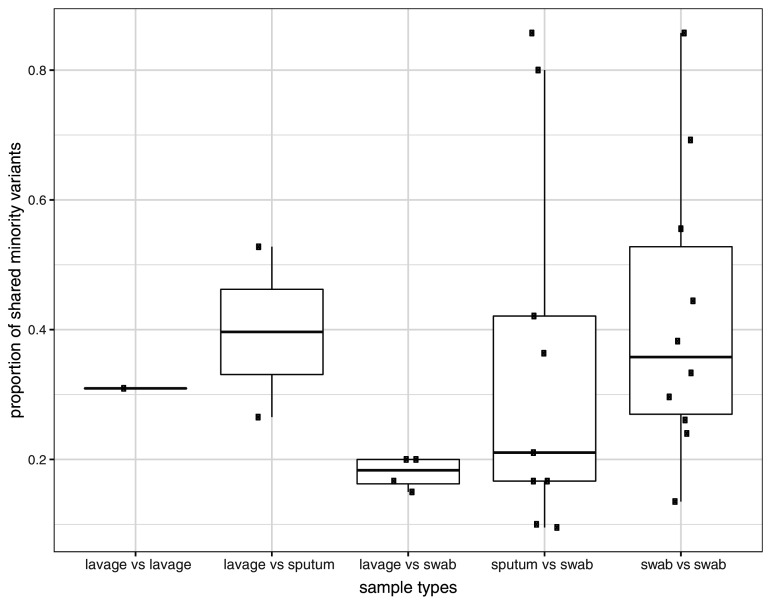
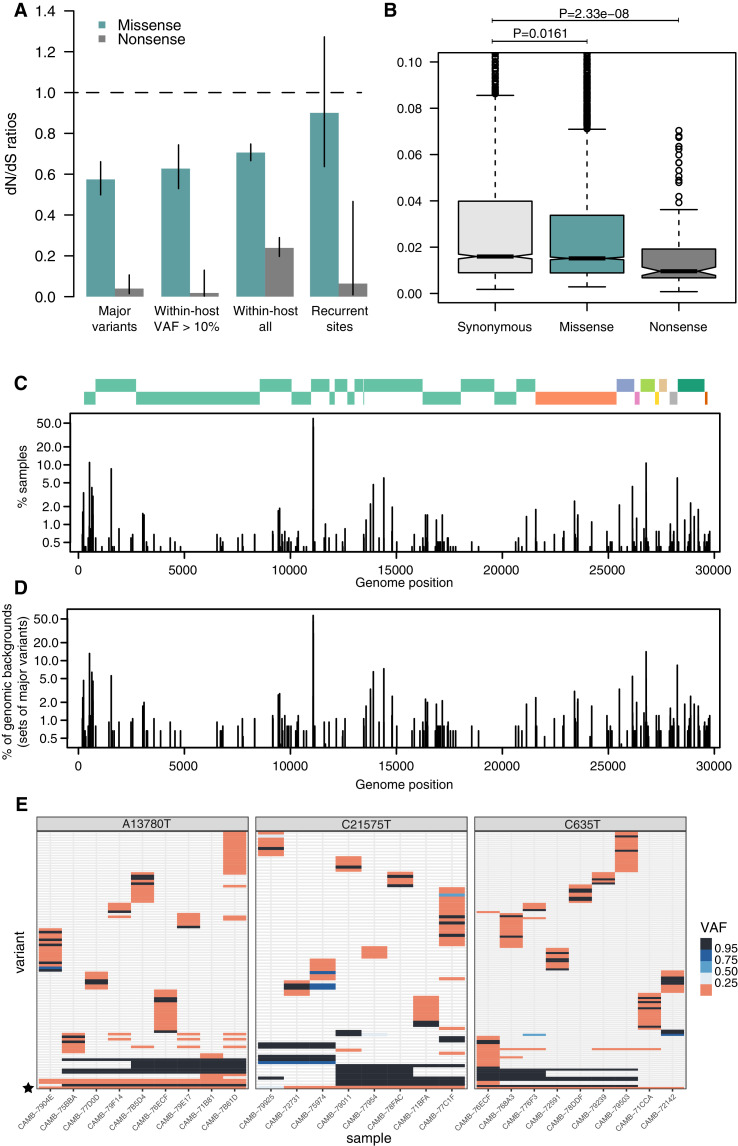
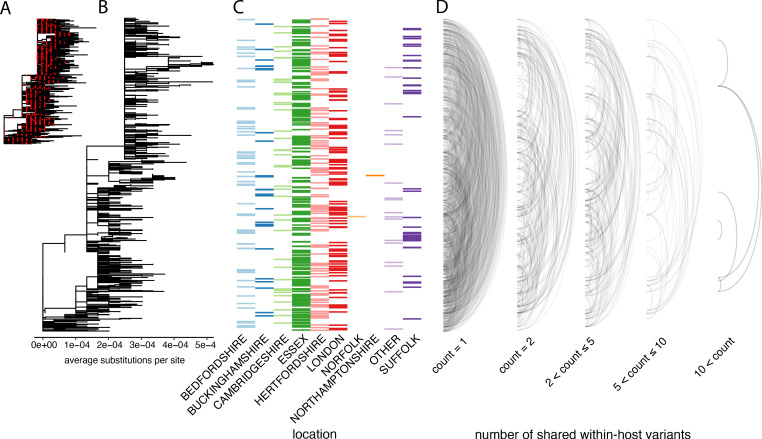
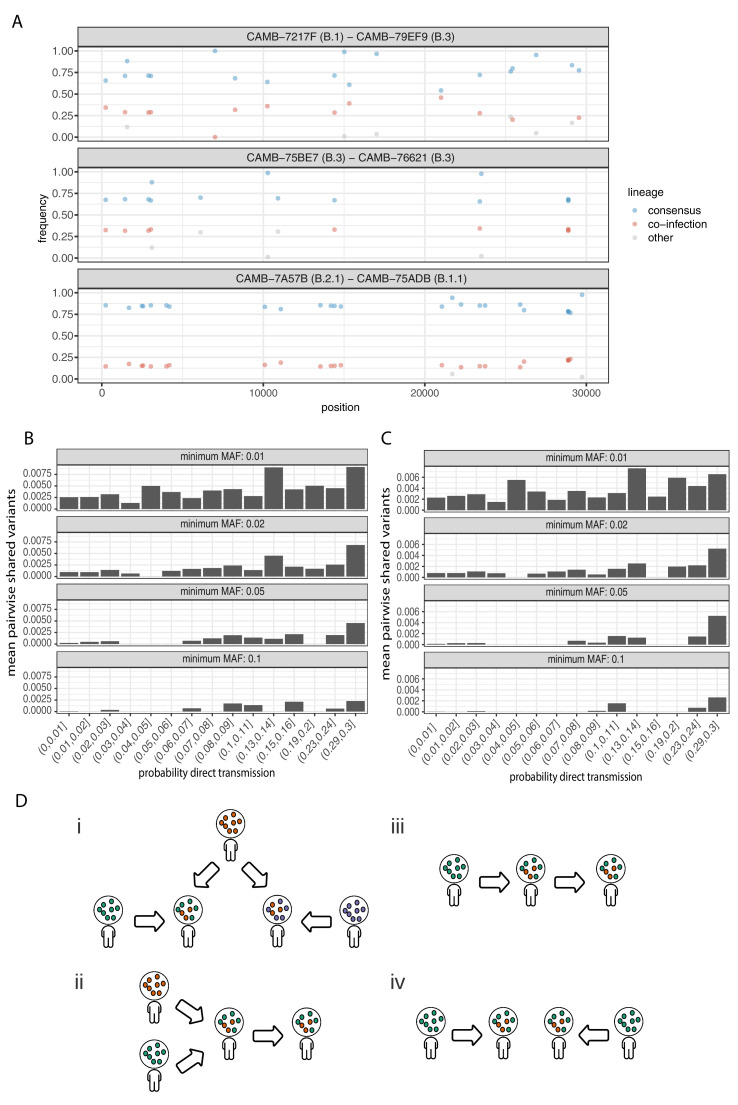
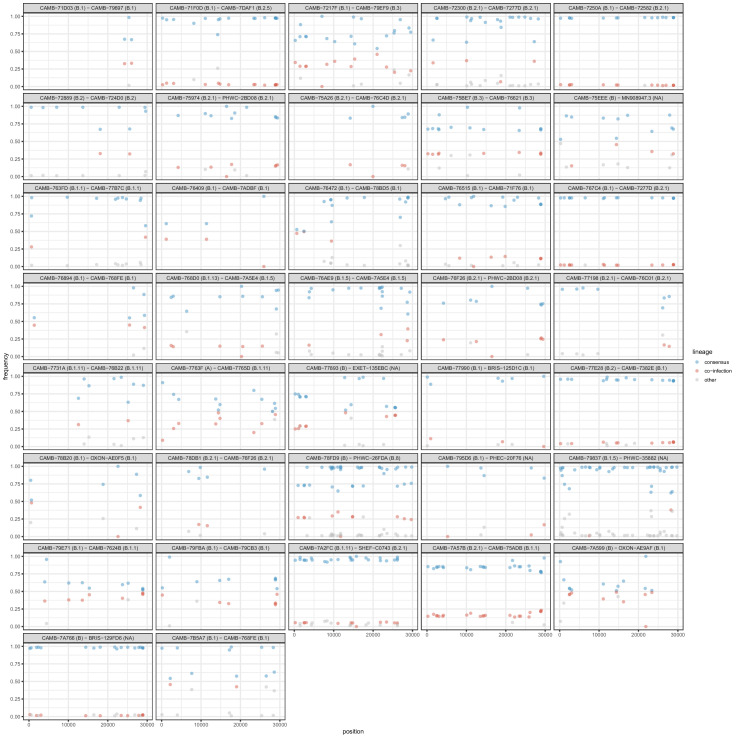
Monitoring the spread of SARS-CoV-2 and reconstructing transmission chains has become a major public health focus for many governments around the world. The modest mutation rate and rapid transmission of SARS-CoV-2 prevents the reconstruction of transmission chains from consensus genome sequences, but within-host genetic diversity could theoretically help identify close contacts. Here we describe the patterns of within-host diversity in 1181 SARS-CoV-2 samples sequenced to high depth in duplicate. 95.1% of samples show within-host mutations at detectable allele frequencies. Analyses of the mutational spectra revealed strong strand asymmetries suggestive of damage or RNA editing of the plus strand, rather than replication errors, dominating the accumulation of mutations during the SARS-CoV-2 pandemic. Within- and between-host diversity show strong purifying selection, particularly against nonsense mutations. Recurrent within-host mutations, many of which coincide with known phylogenetic homoplasies, display a spectrum and patterns of purifying selection more suggestive of mutational hotspots than recombination or convergent evolution. While allele frequencies suggest that most samples result from infection by a single lineage, we identify multiple putative examples of co-infection. Integrating these results into an epidemiological inference framework, we find that while sharing of within-host variants between samples could help the reconstruction of transmission chains, mutational hotspots and rare cases of superinfection can confound these analyses.
Keywords: SARS-CoV-2; epidemiology; genetics; genomics; global health; mutational spectrum; transmission; within-host.
Plain language summary
The COVID-19 pandemic has had major health impacts across the globe. The scientific community has focused much attention on finding ways to monitor how the virus responsible for the pandemic, SARS-CoV-2, spreads. One option is to perform genetic tests, known as sequencing, on SARS-CoV-2 samples to determine the genetic code of the virus and to find any differences or mutations in the genes between the viral samples. Viruses mutate within their hosts and can develop into variants that are able to more easily transmit between hosts. Genetic sequencing can reveal how genetically similar two SARS-CoV-2 samples are. But tracking how SARS-CoV-2 moves from one person to the next through sequencing can be tricky. Even a sample of SARS-CoV-2 viruses from the same individual can display differences in their genetic material or within-host variants. Could genetic testing of within-host variants shed light on factors driving SARS-CoV-2 to evolve in humans? To get to the bottom of this, Tonkin-Hill, Martincorena et al. probed the genetics of SARS-CoV-2 within-host variants using 1,181 samples. The analyses revealed that 95.1% of samples contained within-host variants. A number of variants occurred frequently in many samples, which were consistent with mutational hotspots in the SARS-CoV-2 genome. In addition, within-host variants displayed mutation patterns that were similar to patterns found between infected individuals. The shared within-host variants between samples can help to reconstruct transmission chains. However, the observed mutational hotspots and the detection of multiple strains within an individual can make this challenging. These findings could be used to help predict how SARS-CoV-2 evolves in response to interventions such as vaccines. They also suggest that caution is needed when using information on within-host variants to determine transmission between individuals.
© 2021, Tonkin-Hill et al.
Conflict of interest statement
GT, IM, RA, AL, MG, IJ, DJ, NP, SL, MQ, SG, CA, MS, WH, LM, GH, AJ, YC, MH, MP, IG, AY, LC, SC, TF, FK, CH, MC, SP, AA, RN, EH, JS, SB, JB, MT, IG, CL, DK No competing interests declared
Figures

References
-
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Imielinsk M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdés-Mas R, van Buuren MM, van 't Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR, Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human Cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. - DOI - PMC - PubMed
-
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, Getz G, Rozen SG, Stratton MR, PCAWG Mutational Signatures Working Group. PCAWG Consortium The repertoire of mutational signatures in human Cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. - DOI - PMC - PubMed
-
- Arias A, Watson SJ, Asogun D, Tobin EA, Lu J, Phan MVT, Jah U, Wadoum REG, Meredith L, Thorne L, Caddy S, Tarawalie A, Langat P, Dudas G, Faria NR, Dellicour S, Kamara A, Kargbo B, Kamara BO, Gevao S, Cooper D, Newport M, Horby P, Dunning J, Sahr F, Brooks T, Simpson AJH, Groppelli E, Liu G, Mulakken N, Rhodes K, Akpablie J, Yoti Z, Lamunu M, Vitto E, Otim P, Owilli C, Boateng I, Okoror L, Omomoh E, Oyakhilome J, Omiunu R, Yemisis I, Adomeh D, Ehikhiametalor S, Akhilomen P, Aire C, Kurth A, Cook N, Baumann J, Gabriel M, Wölfel R, Di Caro A, Carroll MW, Günther S, Redd J, Naidoo D, Pybus OG, Rambaut A, Kellam P, Goodfellow I, Cotten M. Rapid outbreak sequencing of ebola virus in sierra Leone identifies transmission chains linked to sporadic cases. Virus Evolution. 2016;2:vew016. doi: 10.1093/ve/vew016. - DOI - PMC - PubMed
-
- Bull M. A Nextflow pipeline for running the ARTIC network’s fieldbioinformatics tools with a focus on ncov2019. AGPL-3.0Github. 2020 https://github.com/connor-lab/ncov2019-artic-nf
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
