

OVarFlow: a resource optimized GATK 4 based Open source Variant calling workFlow
- PMID: 34388963
- PMCID: PMC8361789
- DOI: 10.1186/s12859-021-04317-y
OVarFlow: a resource optimized GATK 4 based Open source Variant calling workFlow
Abstract
Background: The advent of next generation sequencing has opened new avenues for basic and applied research. One application is the discovery of sequence variants causative of a phenotypic trait or a disease pathology. The computational task of detecting and annotating sequence differences of a target dataset between a reference genome is known as "variant calling". Typically, this task is computationally involved, often combining a complex chain of linked software tools. A major player in this field is the Genome Analysis Toolkit (GATK). The "GATK Best Practices" is a commonly referred recipe for variant calling. However, current computational recommendations on variant calling predominantly focus on human sequencing data and ignore ever-changing demands of high-throughput sequencing developments. Furthermore, frequent updates to such recommendations are counterintuitive to the goal of offering a standard workflow and hamper reproducibility over time.
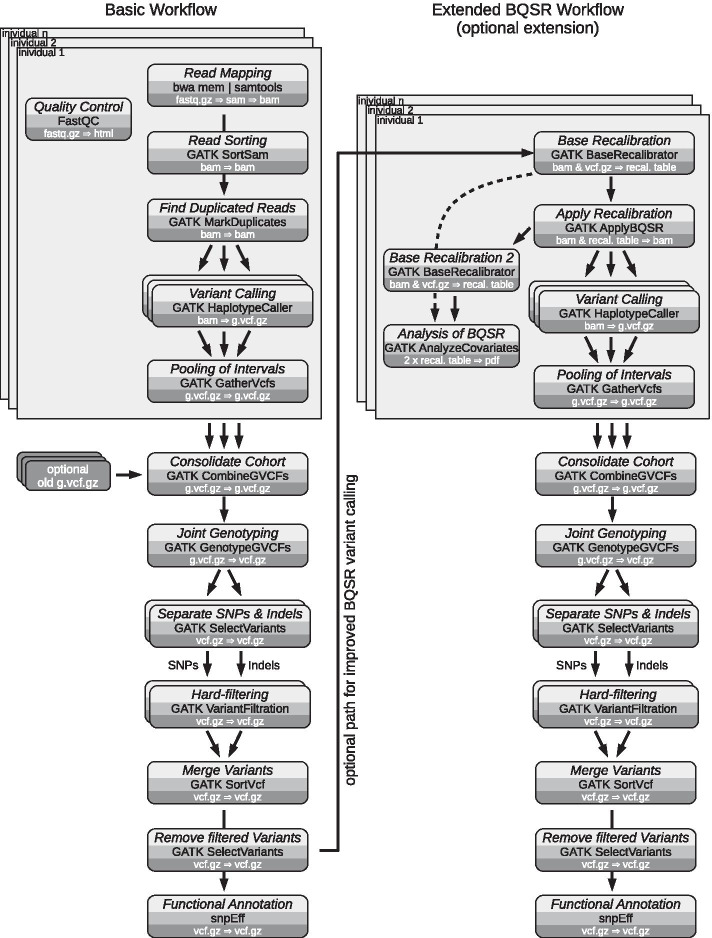
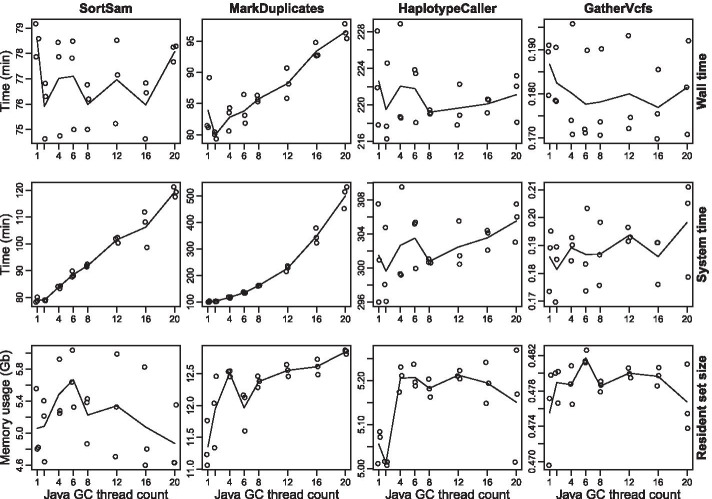
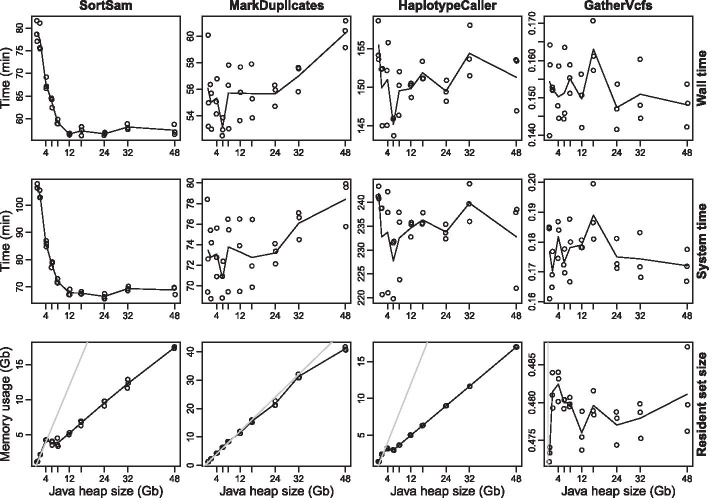
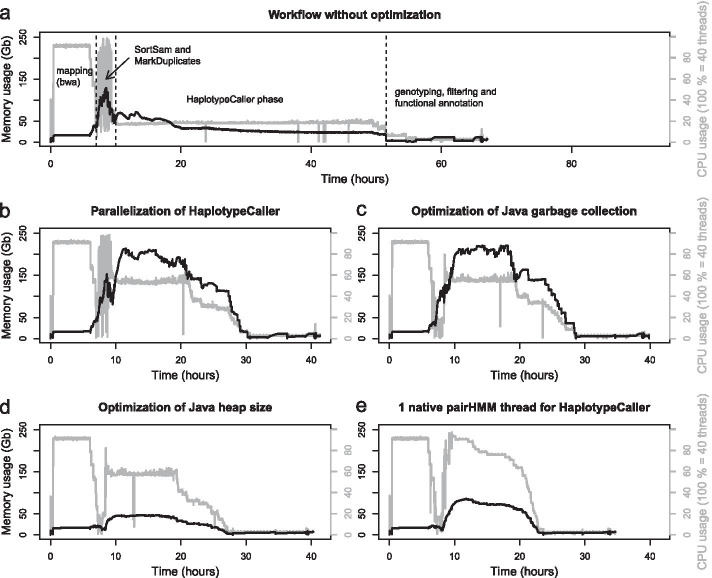
Results: A workflow for automated detection of single nucleotide polymorphisms and insertion-deletions offers a wide range of applications in sequence annotation of model and non-model organisms. The introduced workflow builds on the GATK Best Practices, while enabling reproducibility over time and offering an open, generalized computational architecture. The workflow achieves parallelized data evaluation and maximizes performance of individual computational tasks. Optimized Java garbage collection and heap size settings for the GATK applications SortSam, MarkDuplicates, HaplotypeCaller, and GatherVcfs effectively cut the overall analysis time in half.
Conclusions: The demand for variant calling, efficient computational processing, and standardized workflows is growing. The Open source Variant calling workFlow (OVarFlow) offers automation and reproducibility for a computationally optimized variant calling task. By reducing usage of computational resources, the workflow removes prior existing entry barriers to the variant calling field and enables standardized variant calling.
Keywords: Benchmarking; Data parallelization; GATK; Java; Next generation sequencing; Reproducibility; SNP; Variant calling; indel.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
