

Use of hemagglutinin and neuraminidase amplicon-based high-throughput sequencing with variant analysis to detect co-infection and resolve identical consensus sequences of seasonal influenza in a university setting
- PMID: 34388979
- PMCID: PMC8360813
- DOI: 10.1186/s12879-021-06526-5
Use of hemagglutinin and neuraminidase amplicon-based high-throughput sequencing with variant analysis to detect co-infection and resolve identical consensus sequences of seasonal influenza in a university setting
Abstract
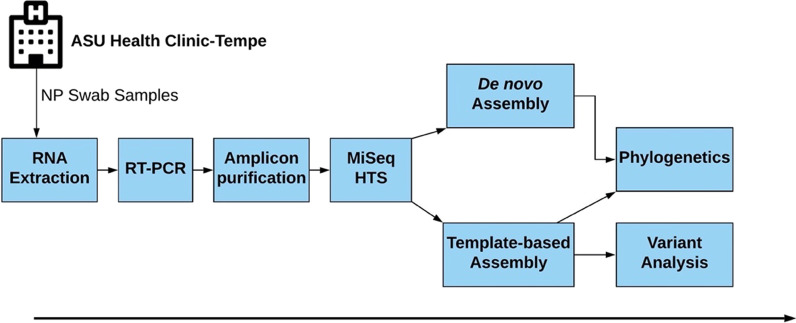
Background: Local transmission of seasonal influenza viruses (IVs) can be difficult to resolve. Here, we study if coupling high-throughput sequencing (HTS) of hemagglutinin (HA) and neuraminidase (NA) genes with variant analysis can resolve strains from local transmission that have identical consensus genome. We analyzed 24 samples collected over four days in January 2020 at a large university in the US. We amplified complete hemagglutinin (HA) and neuraminidase (NA) genomic segments followed by Illumina sequencing. We identified consensus complete HA and NA segments using BLASTn and performed variant analysis on strains whose HA and NA segments were 100% similar.
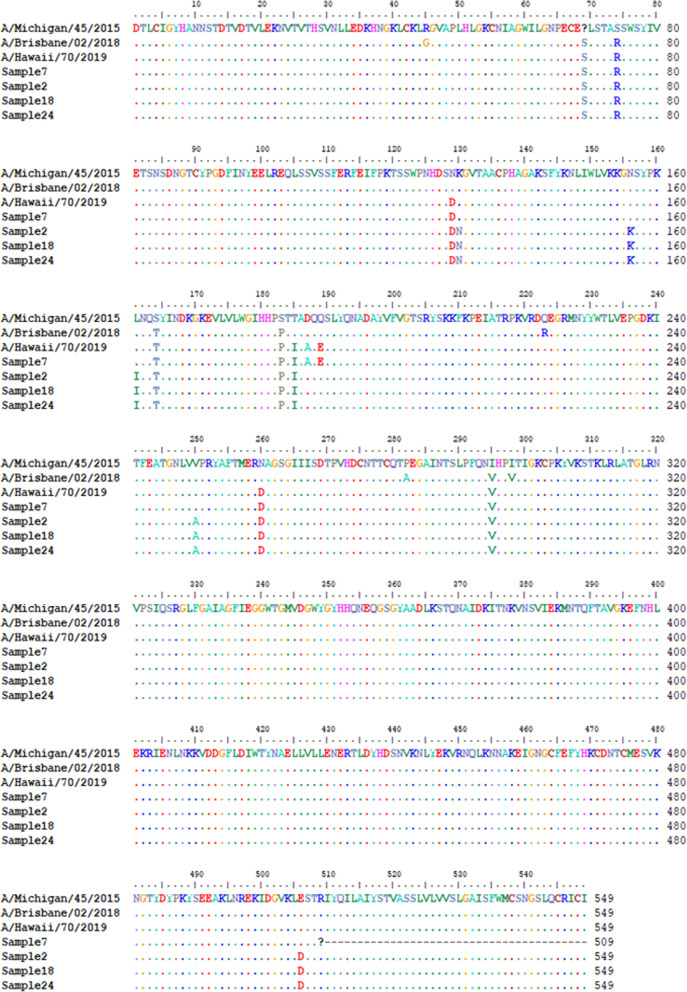
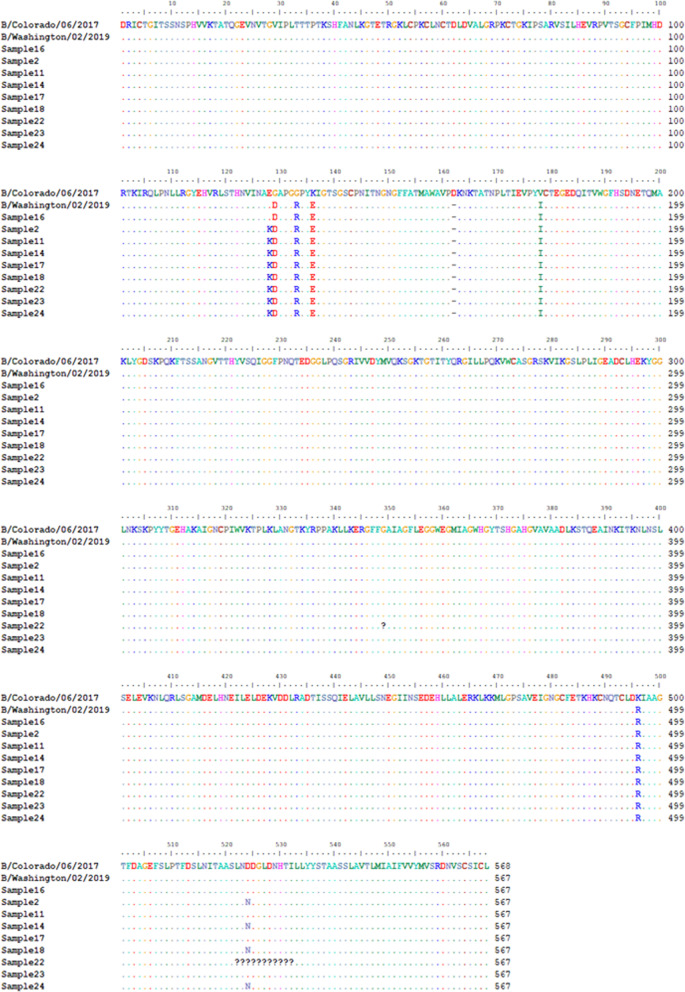
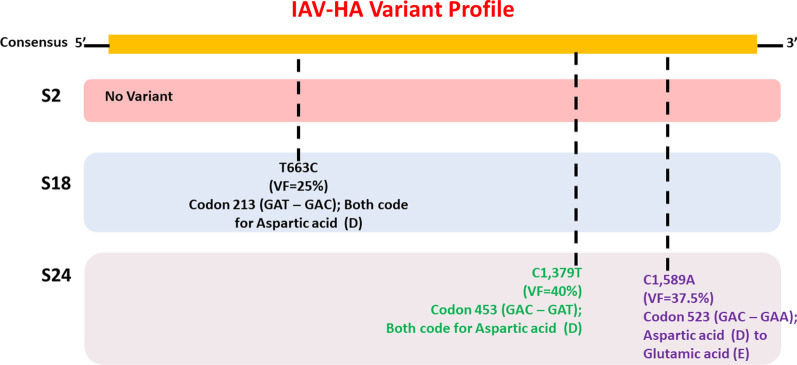
Results: Twelve of the 24 samples were PCR positive, and we detected complete HA and/or NA segments by de novo assembly in 83.33% (10/12) of them. Similarity and phylogenetic analysis showed that 70% (7/10) of the strains were distinct while the remaining 30% had identical consensus sequences. These three samples also had IAV and IBV co-infection. However, subsequent variant analysis showed that they had distinct variant profiles. While the IAV HA of one sample had no variant, another had a T663C mutation and another had both C1379T and C1589A.
Conclusion: In this study, we showed that HTS coupled with variant analysis of only HA and NA genes can help resolve variants that are closely related. We also provide evidence that during a short time period in the 2019-2020 season, co-infection of IAV and IBV occurred on the university campus and both 2020/2021 and 2021/2022 WHO recommended H1N1 vaccine strains were co-circulating.
Keywords: Arizona; HTS; Influenza virus; Local transmission; Orthomyxoviridae; Variant analysis.
© 2021. The Author(s).
Conflict of interest statement
All authors do not have any conflict of interest.
Figures
Similar articles
-
Molecular evolution and characterization of hemagglutinin and neuraminidase of influenza A(H1N1)pdm09 viruses isolated in Beijing, China, during the 2017-2018 and 2018-2019 influenza seasons.Arch Virol. 2021 Jan;166(1):179-189. doi: 10.1007/s00705-020-04869-z. Epub 2020 Nov 3. Arch Virol. 2021. PMID: 33145635
-
Genomic evolution of influenza during the 2023-2024 season, the johns hopkins health system.J Clin Virol. 2024 Oct;174:105718. doi: 10.1016/j.jcv.2024.105718. Epub 2024 Jul 25. J Clin Virol. 2024. PMID: 39079210
-
Phylogenetic and nucleotide sequence analysis of influenza A (H1N1) HA and NA genes of strains isolated from Saudi Arabia.J Infect Dev Ctries. 2017 Jan 30;11(1):81-88. doi: 10.3855/jidc.9259. J Infect Dev Ctries. 2017. PMID: 28141594
-
An overview of influenza A virus genes, protein functions, and replication cycle highlighting important updates.Virus Genes. 2022 Aug;58(4):255-269. doi: 10.1007/s11262-022-01904-w. Epub 2022 Apr 26. Virus Genes. 2022. PMID: 35471490 Review.
-
Influenza virus: Genomic insights, evolution, and its clinical presentation.Microb Pathog. 2025 Aug;205:107671. doi: 10.1016/j.micpath.2025.107671. Epub 2025 May 7. Microb Pathog. 2025. PMID: 40345348 Review.
Cited by
-
Campus-based genomic surveillance uncovers early emergence of a future dominant A(H3N2) influenza clade.medRxiv [Preprint]. 2025 Jun 14:2025.06.13.25329559. doi: 10.1101/2025.06.13.25329559. medRxiv. 2025. PMID: 40585117 Free PMC article. Preprint.
-
The re-emergence of influenza following the COVID-19 pandemic in Victoria, Australia, 2021 to 2022.Euro Surveill. 2023 Sep;28(37):2300118. doi: 10.2807/1560-7917.ES.2023.28.37.2300118. Euro Surveill. 2023. PMID: 37707981 Free PMC article.
-
Influenza B virus from respiratory samples of individuals with influenza-like illness in a University Campus in Arizona USA, in February 2025.Microbiol Resour Announc. 2025 Aug 14;14(8):e0034425. doi: 10.1128/mra.00344-25. Epub 2025 Jul 7. Microbiol Resour Announc. 2025. PMID: 40623801 Free PMC article.
-
IAVCP (Influenza A Virus Consensus and Phylogeny): Automatic Identification of the Genomic Sequence of the Influenza A Virus from High-Throughput Sequencing Data.Viruses. 2024 May 29;16(6):873. doi: 10.3390/v16060873. Viruses. 2024. PMID: 38932165 Free PMC article.
References
-
- Andrews S. FastQC. Babraham Bioinformatics. 2020. www.bioinformatics.babraham.ac.uk/projects/fastqc/.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
