

Evolutionary trajectory of SARS-CoV-2 and emerging variants
- PMID: 34389034
- PMCID: PMC8361246
- DOI: 10.1186/s12985-021-01633-w
Evolutionary trajectory of SARS-CoV-2 and emerging variants
Abstract
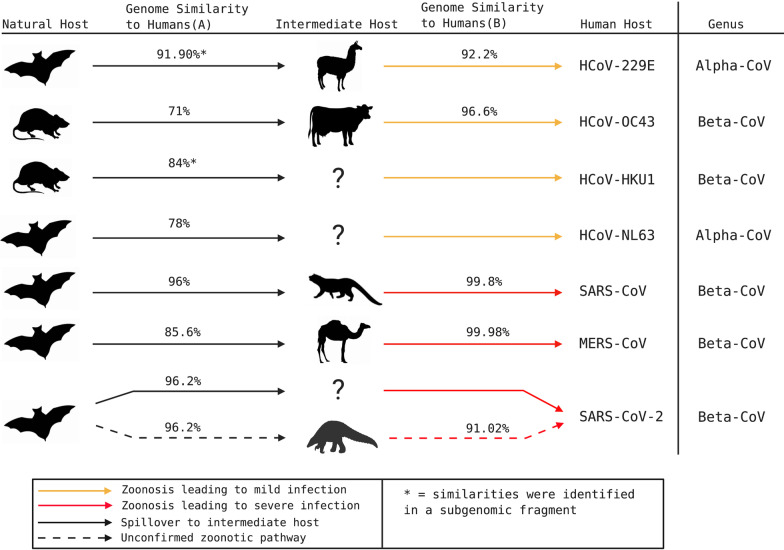
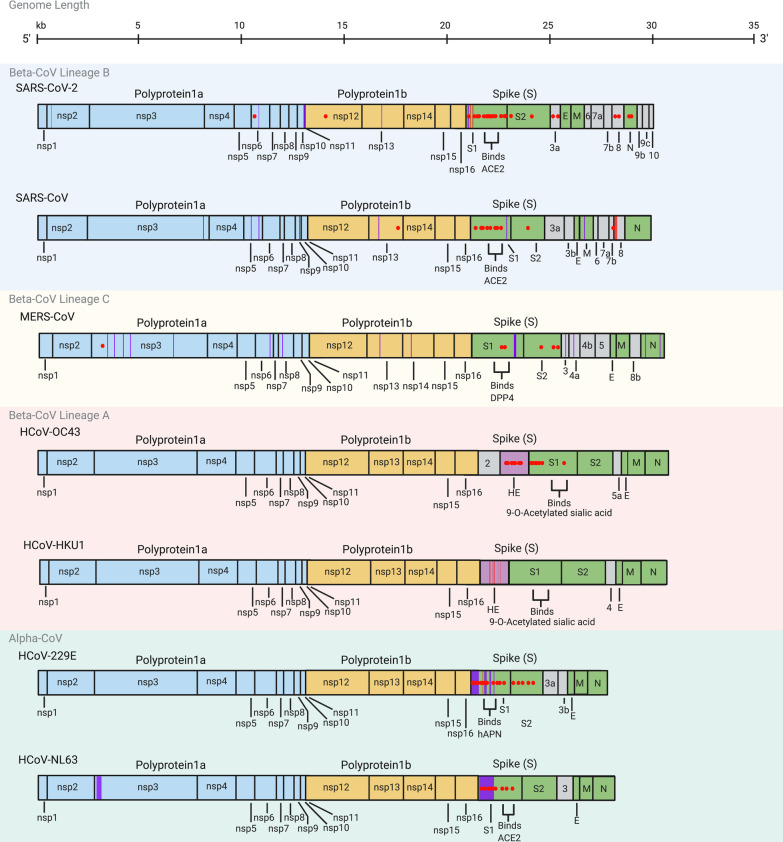
The emergence of a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and more recently, the independent evolution of multiple SARS-CoV-2 variants has generated renewed interest in virus evolution and cross-species transmission. While all known human coronaviruses (HCoVs) are speculated to have originated in animals, very little is known about their evolutionary history and factors that enable some CoVs to co-exist with humans as low pathogenic and endemic infections (HCoV-229E, HCoV-NL63, HCoV-OC43, HCoV-HKU1), while others, such as SARS-CoV, MERS-CoV and SARS-CoV-2 have evolved to cause severe disease. In this review, we highlight the origins of all known HCoVs and map positively selected for mutations within HCoV proteins to discuss the evolutionary trajectory of SARS-CoV-2. Furthermore, we discuss emerging mutations within SARS-CoV-2 and variants of concern (VOC), along with highlighting the demonstrated or speculated impact of these mutations on virus transmission, pathogenicity, and neutralization by natural or vaccine-mediated immunity.
Keywords: Coronavirus; Evolution; Mutations; SARS-CoV-2; Selection; Variants.
© 2021. The Author(s).
Conflict of interest statement
Authors declare no competing interests.
Figures
References
-
- Woo PCY, Lau SKP, Lam CSF, Lau CCY, Tsang AKL, Lau JHN, et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86(7):3995–4008. doi: 10.1128/JVI.06540-11. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
