

Recovery of Deleted Deep Sequencing Data Sheds More Light on the Early Wuhan SARS-CoV-2 Epidemic
- PMID: 34398234
- PMCID: PMC8436388
- DOI: 10.1093/molbev/msab246
Recovery of Deleted Deep Sequencing Data Sheds More Light on the Early Wuhan SARS-CoV-2 Epidemic
Erratum in
-
Correction to: Recovery of Deleted Deep Sequencing Data Sheds More Light on the Early Wuhan SARS-CoV-2 Epidemic.Mol Biol Evol. 2023 Sep 1;40(9):msad201. doi: 10.1093/molbev/msad201. Mol Biol Evol. 2023. PMID: 37772800 Free PMC article. No abstract available.
Abstract
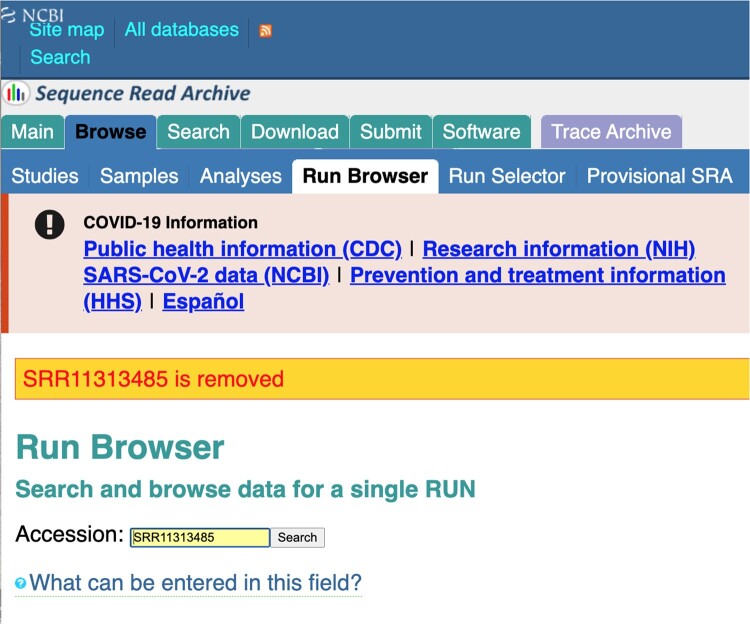
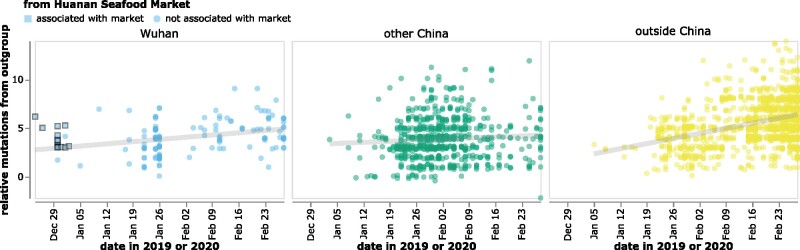
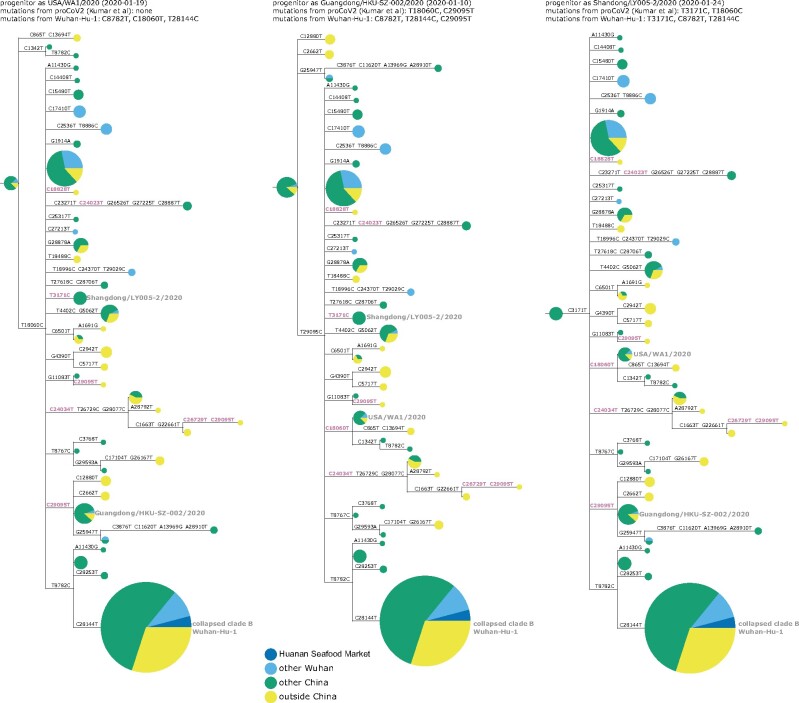
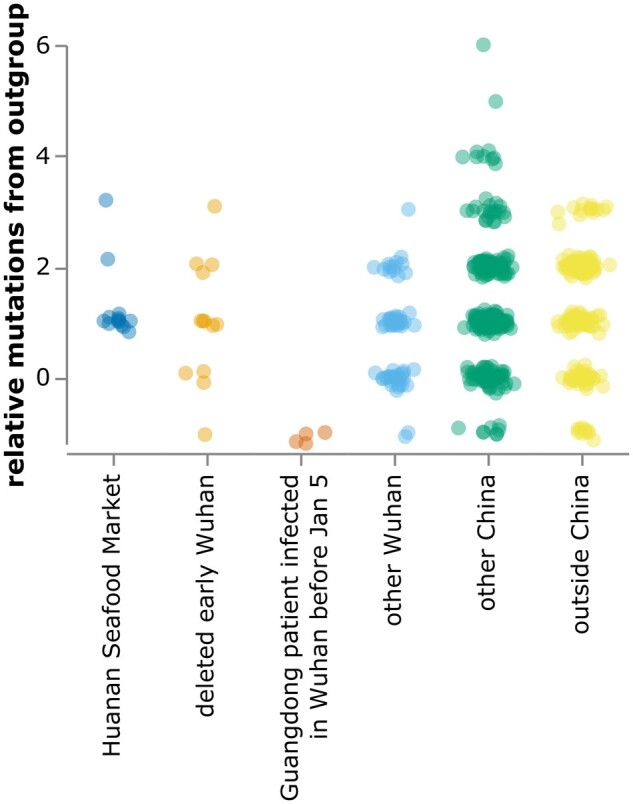
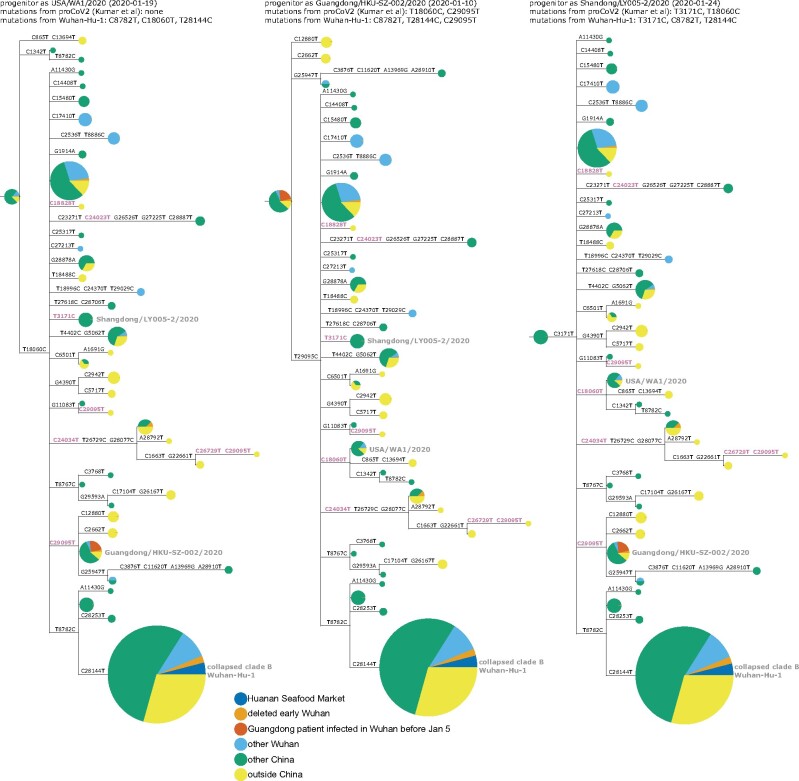
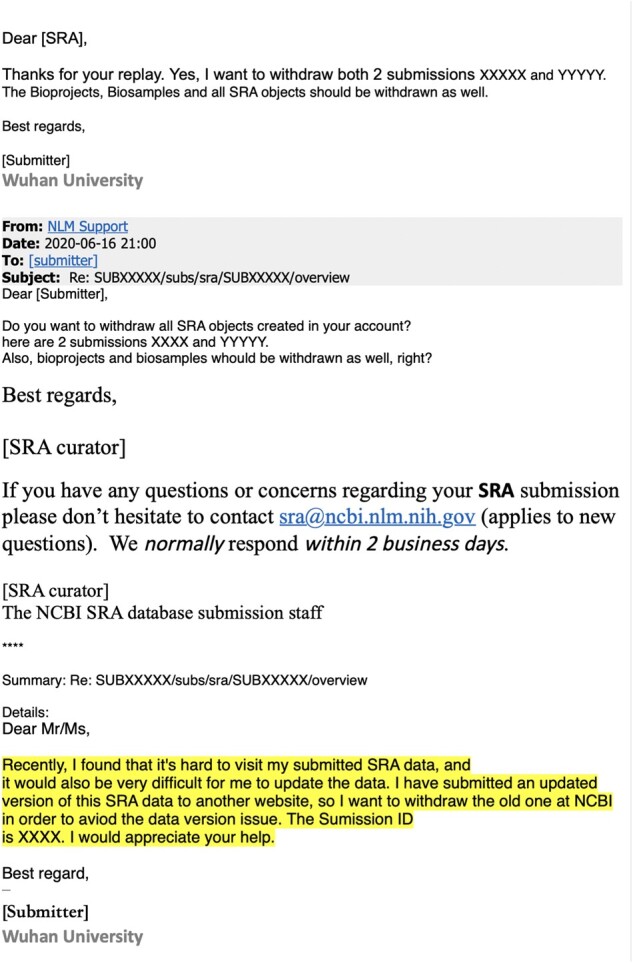
The origin and early spread of SARS-CoV-2 remains shrouded in mystery. Here, I identify a data set containing SARS-CoV-2 sequences from early in the Wuhan epidemic that has been deleted from the NIH's Sequence Read Archive. I recover the deleted files from the Google Cloud and reconstruct partial sequences of 13 early epidemic viruses. Phylogenetic analysis of these sequences in the context of carefully annotated existing data further supports the idea that the Huanan Seafood Market sequences are not fully representative of the viruses in Wuhan early in the epidemic. Instead, the progenitor of currently known SARS-CoV-2 sequences likely contained three mutations relative to the market viruses that made it more similar to SARS-CoV-2's bat coronavirus relatives.
Keywords: COVID-19; SARS-CoV-2; Sequence Read Archive; forensic bioinformatics; phylogenetics.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Conflict of interest statement
The author consults for Moderna on SARS-CoV-2 evolution and epidemiology, consults for Flagship Labs 77 on viral evolution and deep mutational scanning, and has the potential to receive a share of IP revenue as an inventor on a Fred Hutch licensed technology/patent (application WO2020006494) related to deep mutational scanning of viral proteins.
Figures
References
-
- Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW, Castoe TA, Rambaut A, Robertson DL. 2020. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 5(11):1408–1417. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
