

Hidden Viral Sequences in Public Sequencing Data and Warning for Future Emerging Diseases
- PMID: 34399612
- PMCID: PMC8406186
- DOI: 10.1128/mBio.01638-21
Hidden Viral Sequences in Public Sequencing Data and Warning for Future Emerging Diseases
Abstract
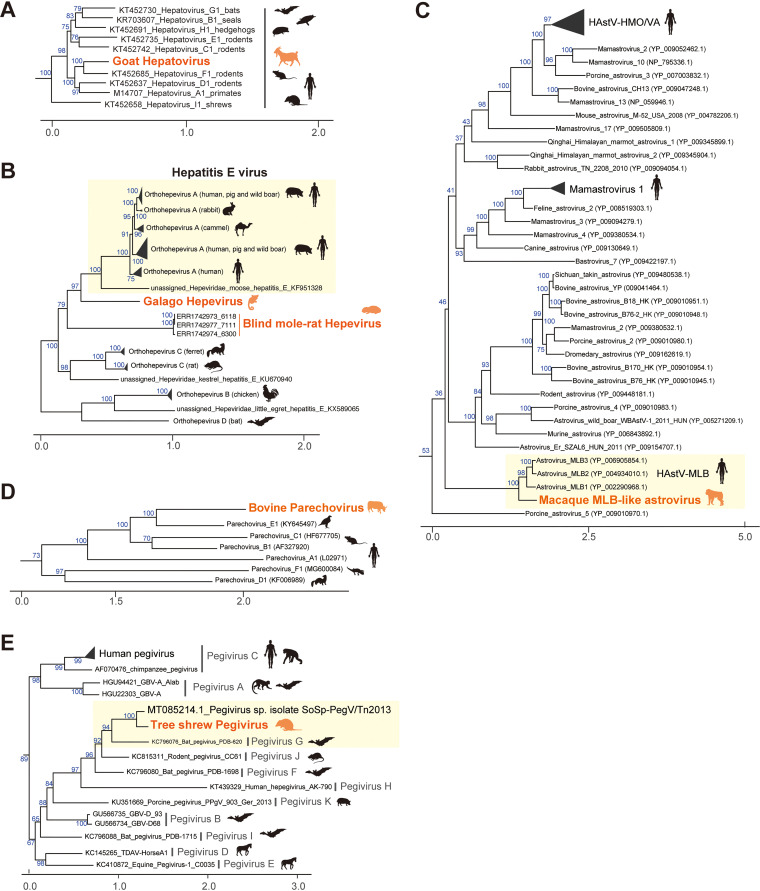
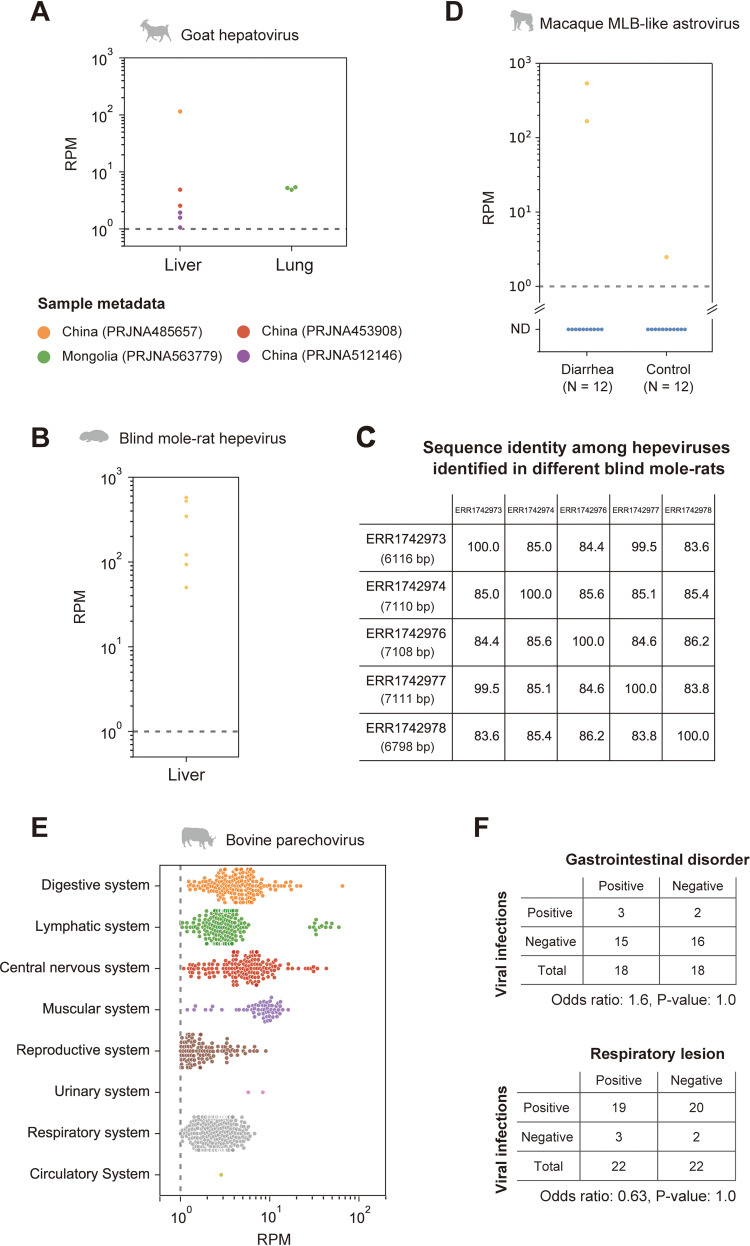
RNA viruses cause numerous emerging diseases, mostly due to transmission from mammalian and avian reservoirs. Large-scale surveillance of RNA viral infections in these animals is a fundamental step for controlling viral infectious diseases. Metagenomic analysis is a powerful method for virus identification with low bias and has contributed substantially to the discovery of novel viruses. Deep-sequencing data have been collected from diverse animals and accumulated in public databases, which can be valuable resources for identifying unknown viral sequences. Here, we screened for infections of 33 RNA viral families in publicly available mammalian and avian sequencing data and found approximately 900 hidden viral infections. We also discovered six nearly complete viral genomes in livestock, wild, and experimental animals: hepatovirus in a goat, hepeviruses in blind mole-rats and a galago, astrovirus in macaque monkeys, parechovirus in a cow, and pegivirus in tree shrews. Some of these viruses were phylogenetically close to human-pathogenic viruses, suggesting the potential risk of causing disease in humans upon infection. Furthermore, infections of five novel viruses were identified in several different individuals, indicating that their infections may have already spread in the natural host population. Our findings demonstrate the reusability of public sequencing data for surveying viral infections and identifying novel viral sequences, presenting a warning about a new threat of viral infectious disease to public health. IMPORTANCE Monitoring the spread of viral infections and identifying novel viruses capable of infecting humans through animal reservoirs are necessary to control emerging viral diseases. Massive amounts of sequencing data collected from various animals are publicly available, and these data may contain sequences originating from a wide variety of viruses. Here, we analyzed more than 46,000 public sequencing data and identified approximately 900 hidden RNA viral infections in mammalian and avian samples. Some viruses discovered in this study were genetically similar to pathogens that cause hepatitis, diarrhea, or encephalitis in humans, suggesting the presence of new threats to public health. Our study demonstrates the effectiveness of reusing public sequencing data to identify known and unknown viral infections, indicating that future continuous monitoring of public sequencing data by metagenomic analyses would help prepare and mitigate future viral pandemics.
Keywords: RNA virus; bioinformatics; molecular epidemiology; public health; virus diversity; zoonosis.
Figures
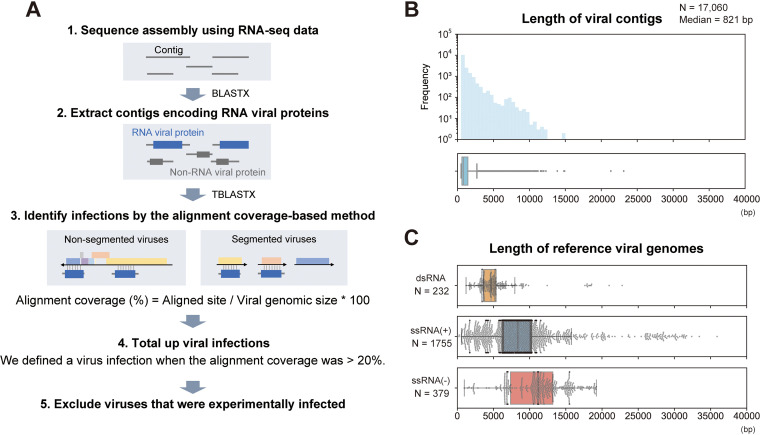
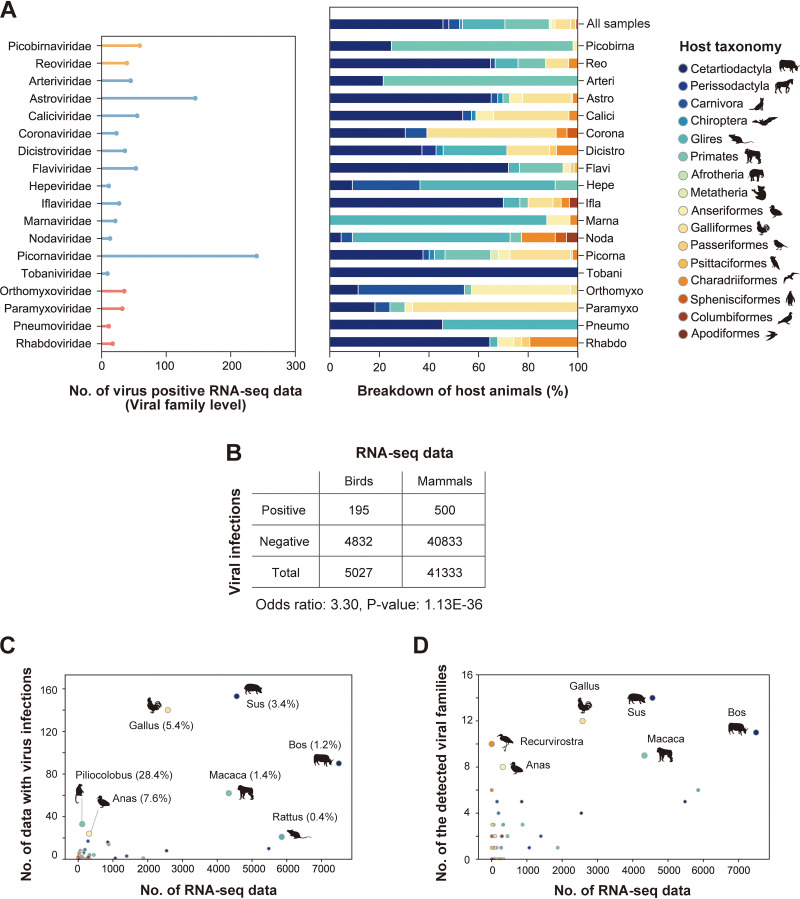
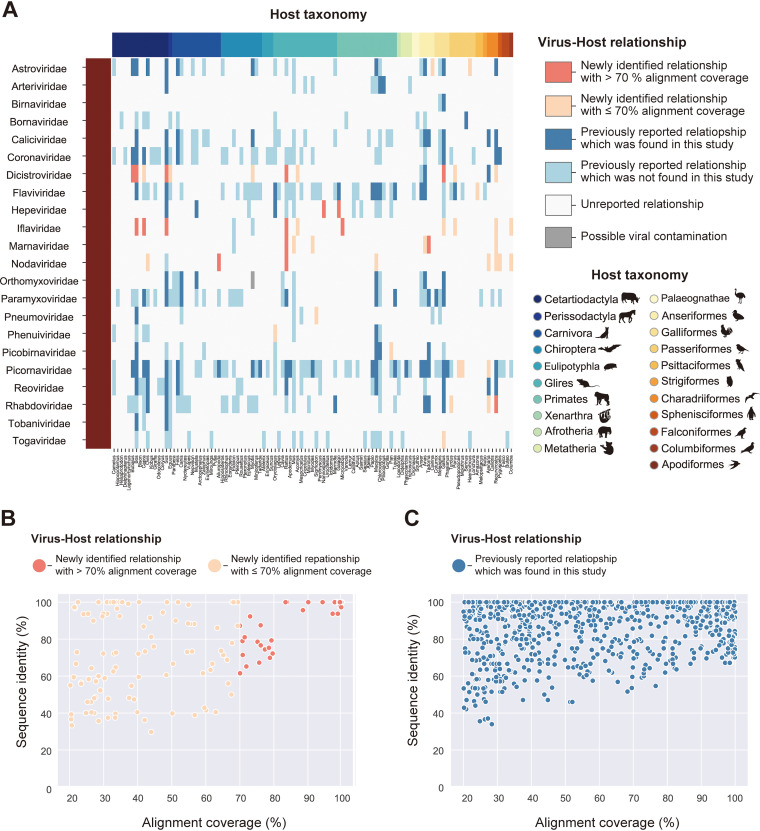
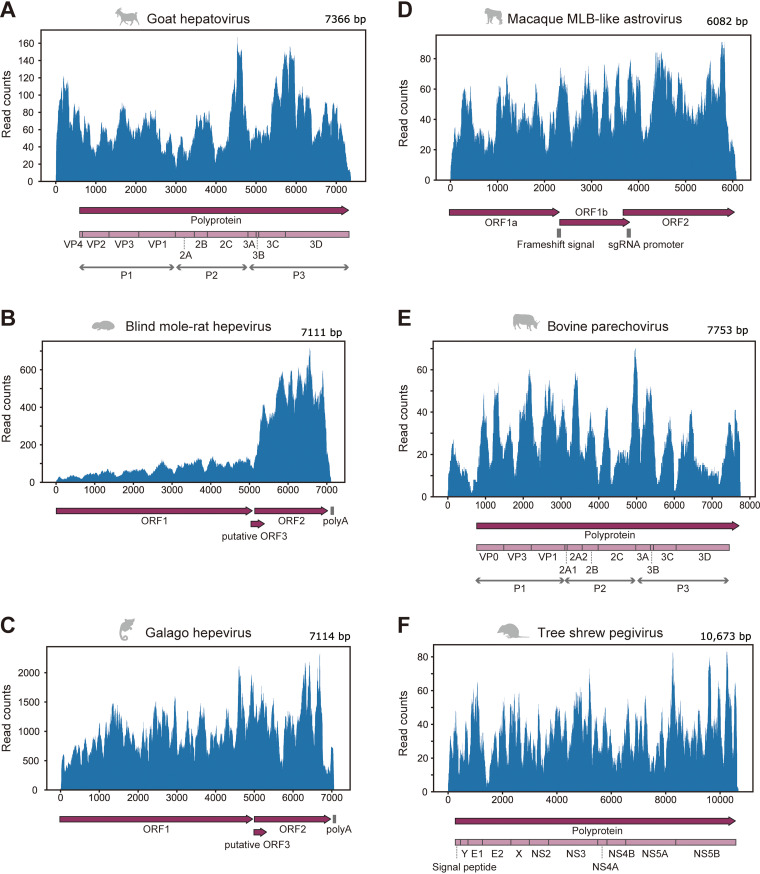
References
-
- Otte M, Nugent R, McLeod A. 2004. Transboundary animal diseases: assessment of socio-economic impacts and institutional responses, p 119–126. Food and Agriculture Organization, Rome, Italy.
