

Chromatin interaction neural network (ChINN): a machine learning-based method for predicting chromatin interactions from DNA sequences
- PMID: 34399797
- PMCID: PMC8365954
- DOI: 10.1186/s13059-021-02453-5
Chromatin interaction neural network (ChINN): a machine learning-based method for predicting chromatin interactions from DNA sequences
Abstract
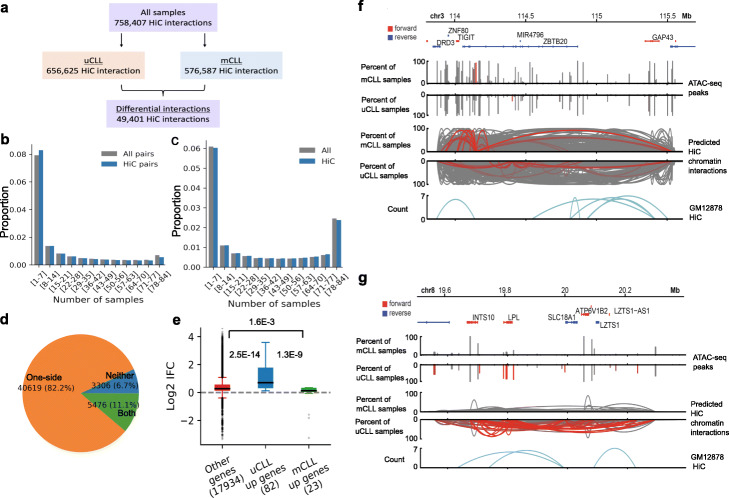
Chromatin interactions play important roles in regulating gene expression. However, the availability of genome-wide chromatin interaction data is limited. We develop a computational method, chromatin interaction neural network (ChINN), to predict chromatin interactions between open chromatin regions using only DNA sequences. ChINN predicts CTCF- and RNA polymerase II-associated and Hi-C chromatin interactions. ChINN shows good across-sample performances and captures various sequence features for chromatin interaction prediction. We apply ChINN to 6 chronic lymphocytic leukemia (CLL) patient samples and a published cohort of 84 CLL open chromatin samples. Our results demonstrate extensive heterogeneity in chromatin interactions among CLL patient samples.
Keywords: 3D genome organization; Bioinformatics; ChIA-PET; Chromatin interactions; DNA sequence; Hi-C; Leukemia; Machine learning.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures







References
-
- Schottenfeld D, JDaly JM. Gastrointesinal cancer: epidemiology. In: Kelsen DP, Levin B, Kern SE, Tepper JE, editors. Gastrointestinal oncology: principles and practice. Philadelphia: Lippincott Williams and Wilkins; 2002.
-
- Akıncılar SC, Ekta K, Boon PLS, Unal B, Fullwood MJ, Tergaonkar V. Long-range chromatin interactions drive mutant TERT promoter activation. Cancer Disc. 2016;6(11):1276–1291. doi: 10.1158/2159-8290.CD-16-0177. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
