

Cell type-selective targeted delivery of a recombinant lysosomal enzyme for enzyme therapies
- PMID: 34400331
- PMCID: PMC8636175
- DOI: 10.1016/j.ymthe.2021.08.020
Cell type-selective targeted delivery of a recombinant lysosomal enzyme for enzyme therapies
Abstract
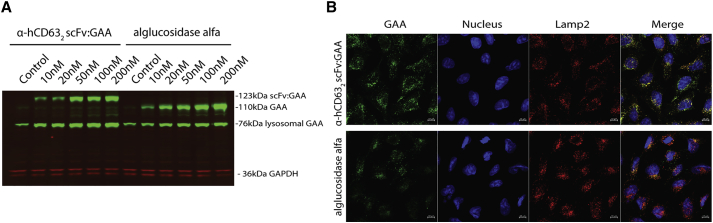
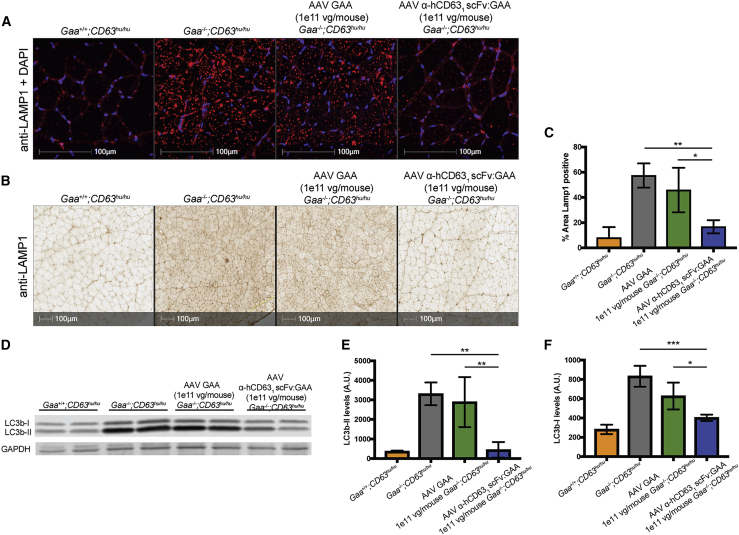
Lysosomal diseases are a class of genetic disorders predominantly caused by loss of lysosomal hydrolases, leading to lysosomal and cellular dysfunction. Enzyme replacement therapy (ERT), where recombinant enzyme is given intravenously, internalized by cells, and trafficked to the lysosome, has been applied to treat several lysosomal diseases. However, current ERT regimens do not correct disease phenotypes in all affected organs because the biodistribution of enzyme uptake does not match that of the affected cells that require the enzyme. We present here targeted ERT, an approach that utilizes antibody-enzyme fusion proteins to target the enzyme to specific cell types. The antibody moiety recognizes transmembrane proteins involved in lysosomal trafficking and that are also preferentially expressed in those cells most affected in disease. Using Pompe disease (PD) as an example, we show that targeted ERT is superior to ERT in treating the skeletal muscle phenotypes of PD mice both as a protein replacement therapeutic and as a gene therapy.
Keywords: enzyme therapy; genetic therapy; glycogen storage disease II; hydrolases; lysosomes; protein transport.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests All authors were employees of Regeneron Pharmaceuticals, Inc., while engaged in the study and may hold stock and/or stock options in the company. A.D.B. and K.D.C. have patent applications for internalizing enzymes and uses thereof.
Figures





References
-
- Hirschhorn R., Reuser A. In: Scriver C.R., Bly W.S., Childs B., Beaudet A.L., Valle D., Kinzler K.W., Vogelstein B., editors. McGraw-Hill; New York: 2000. Glycogen Storage Disease Type II: Acid Alpha-glucosidase (acid maltase) Deficiency; pp. 3389–3420. (The Metabolic and Molecular Bases of Inherited Disease).
-
- Chien Y.-H., Lee N.-C., Chen C.-A., Tsai F.-J., Tsai W.-H., Shieh J.-Y., Huang H.-J., Hsu W.-C., Tsai T.-H., Hwu W.-L. Long-Term Prognosis of Patients with Infantile-Onset Pompe Disease Diagnosed by Newborn Screening and Treated since Birth. J. Pediat. 2015;166:985–991. e1-2. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
