

Combined epidemiological and genomic analysis of nosocomial SARS-CoV-2 infection early in the pandemic and the role of unidentified cases in transmission
- PMID: 34400345
- PMCID: PMC8361005
- DOI: 10.1016/j.cmi.2021.07.040
Combined epidemiological and genomic analysis of nosocomial SARS-CoV-2 infection early in the pandemic and the role of unidentified cases in transmission
Abstract
Objectives: To analyse nosocomial transmission in the early stages of the coronavirus 2019 (COVID-19) pandemic at a large multisite healthcare institution. Nosocomial incidence is linked with infection control interventions.
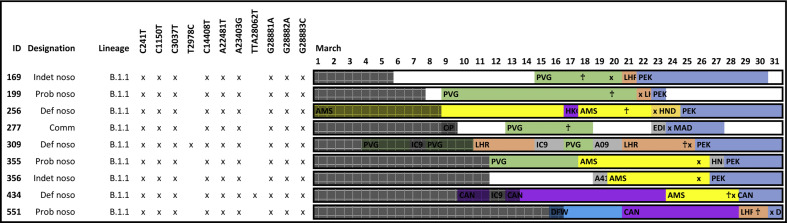
Methods: Viral genome sequence and epidemiological data were analysed for 574 consecutive patients, including 86 nosocomial cases, with a positive PCR test for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) during the first 19 days of the pandemic.
Results: Forty-four putative transmission clusters were found through epidemiological analysis; these included 234 cases and all 86 nosocomial cases. SARS-CoV-2 genome sequences were obtained from 168/234 (72%) of these cases in epidemiological clusters, including 77/86 nosocomial cases (90%). Only 75/168 (45%) of epidemiologically linked, sequenced cases were not refuted by applying genomic data, creating 14 final clusters accounting for 59/77 sequenced nosocomial cases (77%). Viral haplotypes from these clusters were enriched 1-14x (median 4x) compared to the community. Three factors implicated unidentified cases in transmission: (a) community-onset or indeterminate cases were absent in 7/14 clusters (50%), (b) four clusters (29%) had additional evidence of cryptic transmission, and (c) in three clusters (21%) diagnosis of the earliest case was delayed, which may have facilitated transmission. Nosocomial cases decreased to low levels (0-2 per day) despite continuing high numbers of admissions of community-onset SARS-CoV-2 cases (40-50 per day) and before the impact of introducing universal face masks and banning hospital visitors.
Conclusion: Genomics was necessary to accurately resolve transmission clusters. Our data support unidentified cases-such as healthcare workers or asymptomatic patients-as important vectors of transmission. Evidence is needed to ascertain whether routine screening increases case ascertainment and limits nosocomial transmission.
Keywords: Healthcare-associated infection; Molecular epidemiology; Nosocomial transmission; SARS-CoV-2; Whole-genome sequencing.
Copyright © 2021. Published by Elsevier Ltd.
Figures



References
-
- John Hopkins University . 2020. COVID-19 map-johns hopkins coronavirus resource center.https://coronavirus.jhu.edu/map.html
-
- Birrell P., Blake J., van Leeuwen E. 2020. COVID-19: nowcast and forecast.https://joshuablake.github.io/public-RTM-reports/iframe.html
-
- Coronavirus (COVID-19) in the UK. GOV.uk; 2020. https://coronavirus.data.gov.uk/healthcare n.d.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
