

Large-scale and high-resolution mass spectrometry-based proteomics profiling defines molecular subtypes of esophageal cancer for therapeutic targeting
- PMID: 34400640
- PMCID: PMC8368010
- DOI: 10.1038/s41467-021-25202-5
Large-scale and high-resolution mass spectrometry-based proteomics profiling defines molecular subtypes of esophageal cancer for therapeutic targeting
Abstract
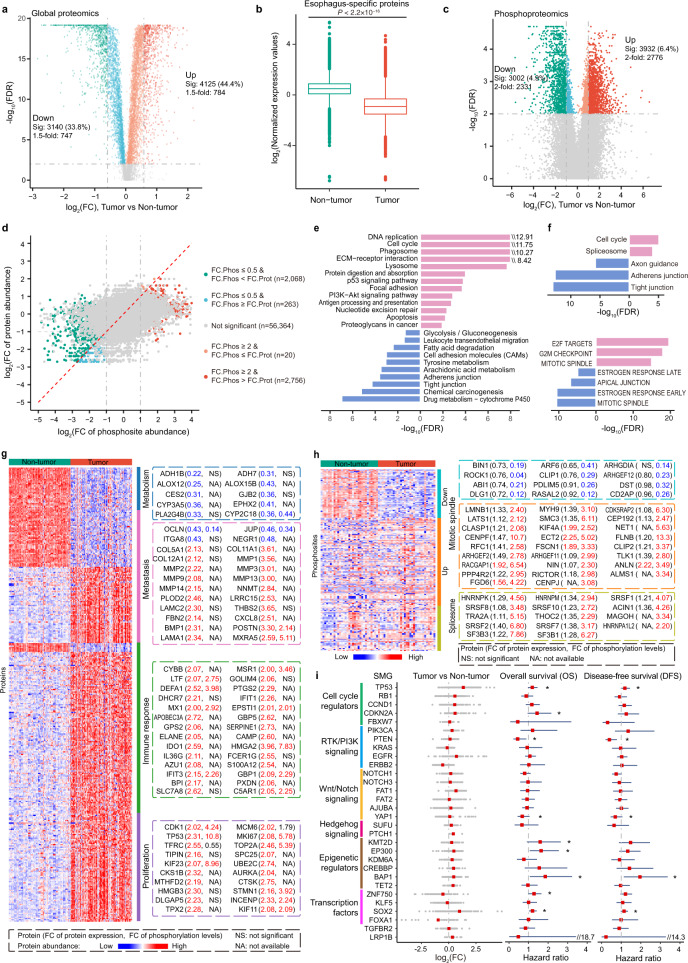
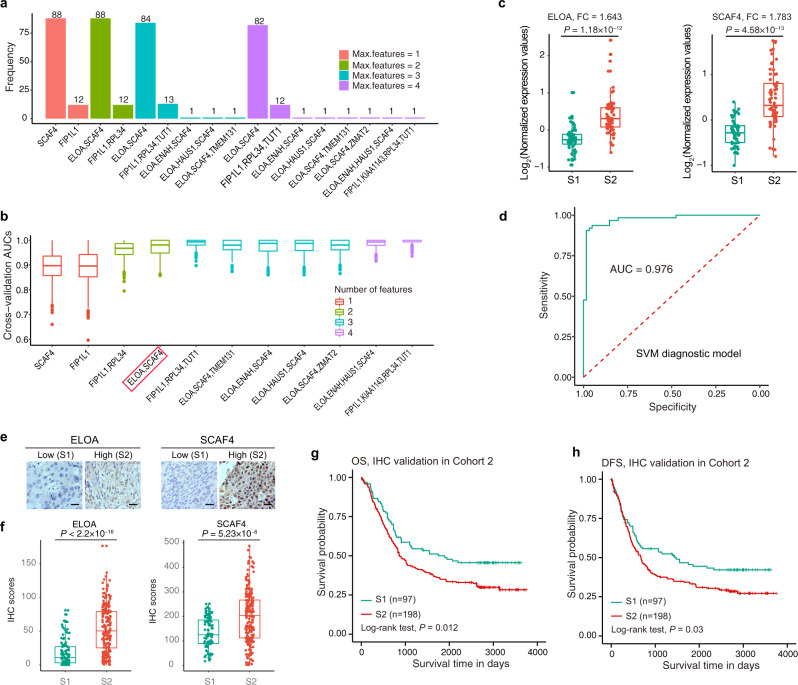
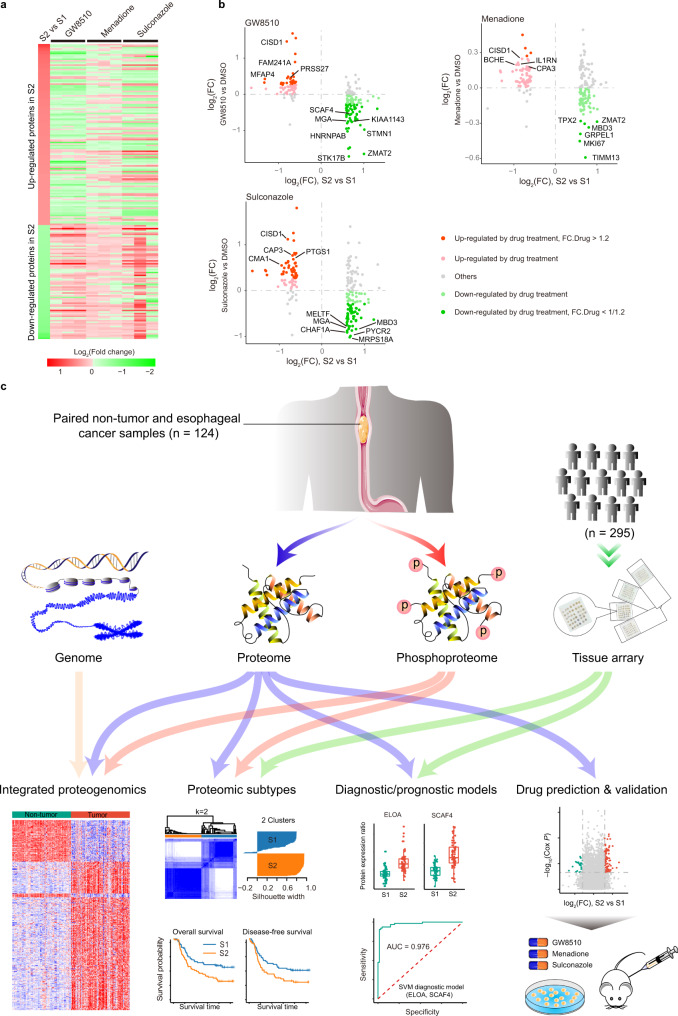
Esophageal cancer (EC) is a type of aggressive cancer without clinically relevant molecular subtypes, hindering the development of effective strategies for treatment. To define molecular subtypes of EC, we perform mass spectrometry-based proteomic and phosphoproteomics profiling of EC tumors and adjacent non-tumor tissues, revealing a catalog of proteins and phosphosites that are dysregulated in ECs. The EC cohort is stratified into two molecular subtypes-S1 and S2-based on proteomic analysis, with the S2 subtype characterized by the upregulation of spliceosomal and ribosomal proteins, and being more aggressive. Moreover, we identify a subtype signature composed of ELOA and SCAF4, and construct a subtype diagnostic and prognostic model. Potential drugs are predicted for treating patients of S2 subtype, and three candidate drugs are validated to inhibit EC. Taken together, our proteomic analysis define molecular subtypes of EC, thus providing a potential therapeutic outlook for improving disease outcomes in patients with EC.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
